

This is part four of a story on utilizing Y-DNA tests to gain knowledge or leads on the patrilineal line of the Griff(is)(es)(ith) family. This part of the story focuses on the analysis of Y-STR test results to possibly locate genetic ancestors.
Working with Y-STRs (and Y-SNPs) and the various types of tests and Y-DNA tools requires covering the topics of genetic distance, modal haplotypes, ancestral haplotypes and the Most Recent Common Ancestor.
Most Common Ancestor: A Peculiar Concept
A number of genetic studies argue that all humans are related genealogically to each other over what can be considered as surprisingly short time scales. [1] Few of us have knowledge of family histories more than a few generations back. Moreover, these ancestors often do not contribute any genetic material to us [2] .

In 2004 mathematical modeling and computer simulations by a group of statisticians indicated that our most recent common ancestor probably lived no earlier than 1400 B.C. and possibly as recently as A.D. 55. Additional simulations, taking into account the geographical separation of continents and islands and less random patterns of mating in real life suggest that some populations are separated by up to a few thousand years, with a most recent common ancestor perhaps 76 generations back (about 336 BCE), though some highly remote populations may have been isolated for somewhat longer [3]
The most recent common ancestor of a group of men and the most common ancestor of man are concepts used in genetic genealogy. Their definition and explanation are not entirely intuitive. They can be difficult to comprehend and what do they actually mean. For most of us it is a bit difficult to accept or even comprehend concepts that rest on mathematics or statistics and not hard data. Archaeologists, genealogists, or historians will never uncover ancient artifacts or documentation that identify your most recent common ancestor.
The idea of a genealogical common ancestor resists attempts to demonstrate its existence with a genetic, DNA equivalent. As special as either of ‘these recent individuals’ are within our genealogy, it is very likely that most living people have inherited no DNA from these individuals at all.
This may seem like a paradox: a genealogical ancestor of everybody, from whom most of us have inherited no DNA. It reminds us that genetic and genealogical relationships are different from each other. Many close genealogical relatives are nonetheless genetically and culturally very different from each other. Fifth cousins are not far apart genealogically, but they sometimes share no DNA from their common genealogical ancestors at all. [4]
The following video provides an excellent overview of the interplay between different concepts of genealogy and their implications. The video also touches on the concept of common ancestor, identical ancestors point (IAP), or all common ancestors (ACA) point, or genetic isopoint, and the most recent ancestor. [5]
Genetic Distance
While I brought up the concept of most common ancestor for discussion, our main concern is really with something that is more manageable to comprehend in terms of genetic distance: genetic distance based on the most recent common ancestor. It still might be confusing but not mind blowing.
Genetic distance, is a concept used more as an operational concept by FamilyTree DNA (FTDNA). It is a concept that ranks individual test kits according to how close they appear to be to each other based on the number of allele differences on designated short tandem repeats (STRs).
Genetic distance can also be calculated using Single-nucleotide polymorphisms (SNPs) by comparing the time distance between different haplogroup branches. For the most part the concept is used in the context of comparing genetic test results between two or more Y-STR test kits to determine if they are genetically ‘closely related’. [6]
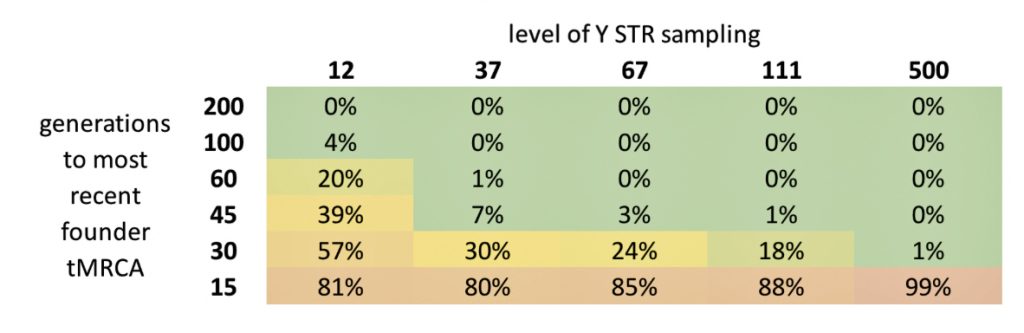
Genetic distance is based on the analysis of STR data, is the result of calculating the number of mutation events which have occurred between two or more individuals in their respective haplotypes. The more STR’s sampled and compared, the more reliable is the estimate of genetic distance.
Most Common Recent Ancestor
In genetic genealogy, the most recent common ancestor (tMRCA) of any set of individuals is the most recent individual from which all the people in the group are directly descended. [7] Estimating TMRCAs is not an exact science. Because it is not an exact science, questions and answers regarding TMRCA should be phased in general terms. For example, is the MRCA likely to be within the time of surnames or is the MRCA more likely to be in the 1`700’s or the 1600’s. Generally, TMRCA concept can be used to give a working theory or hypothesis about which general time frame the common ancestor may have lived.
The results of various type of analyses that calculate genetic distance will point to the most recent common ancestor of a group of men.
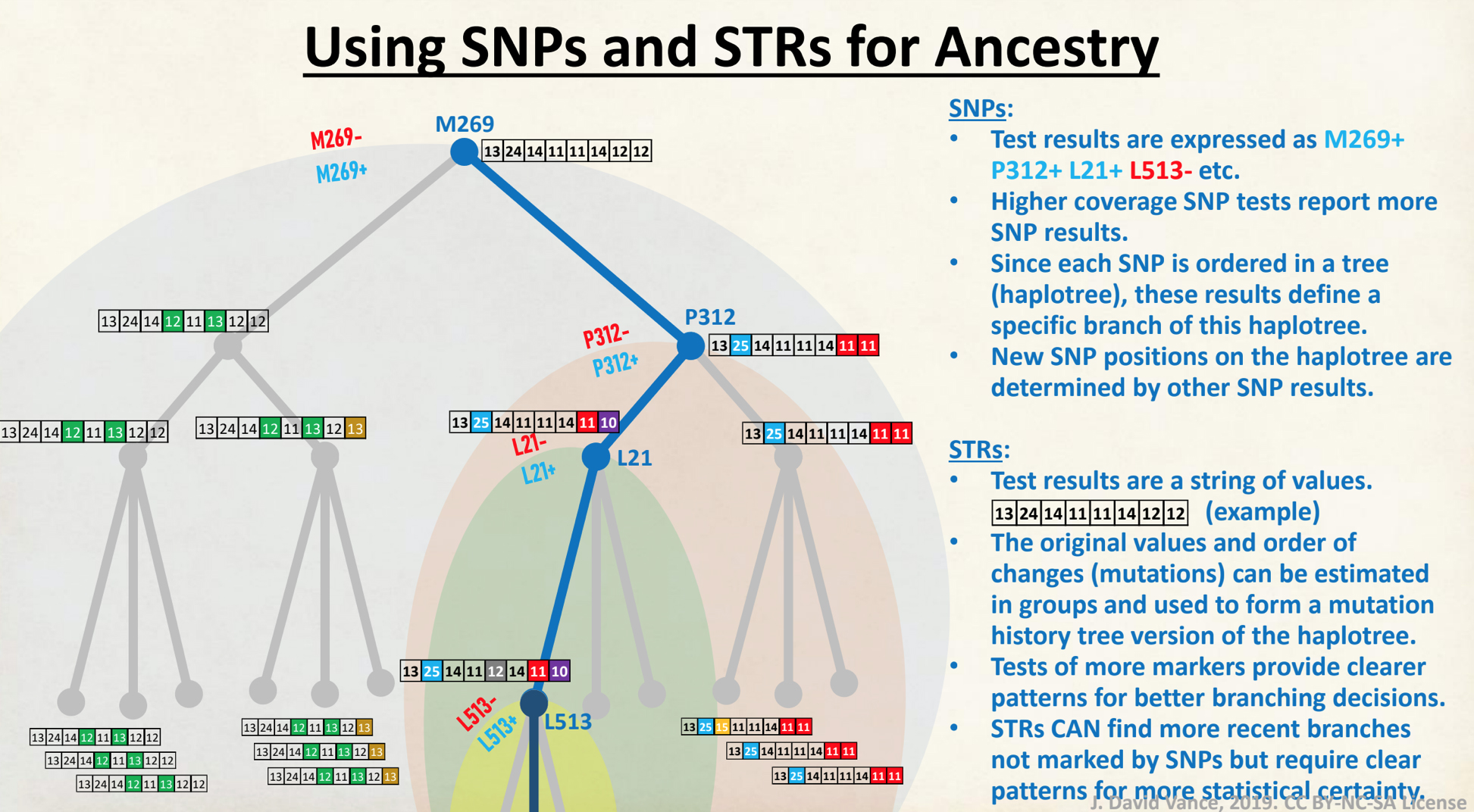
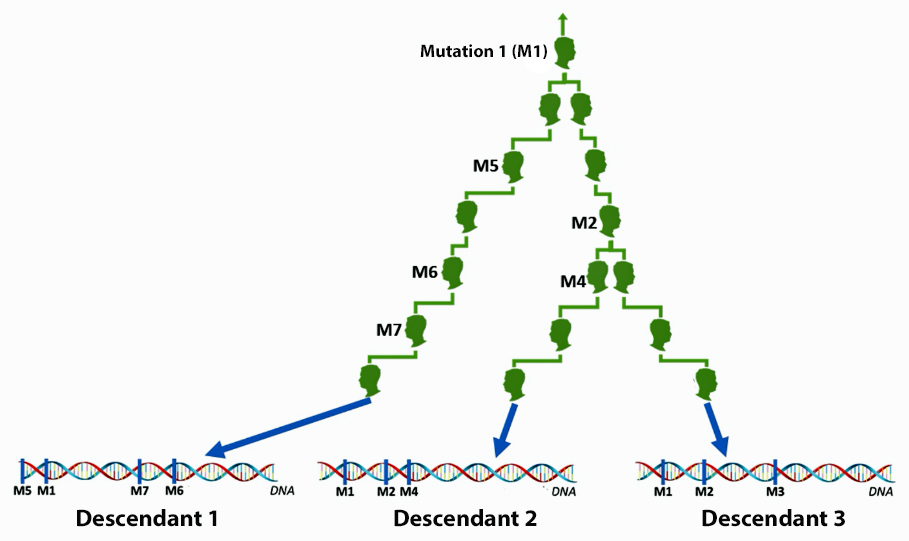
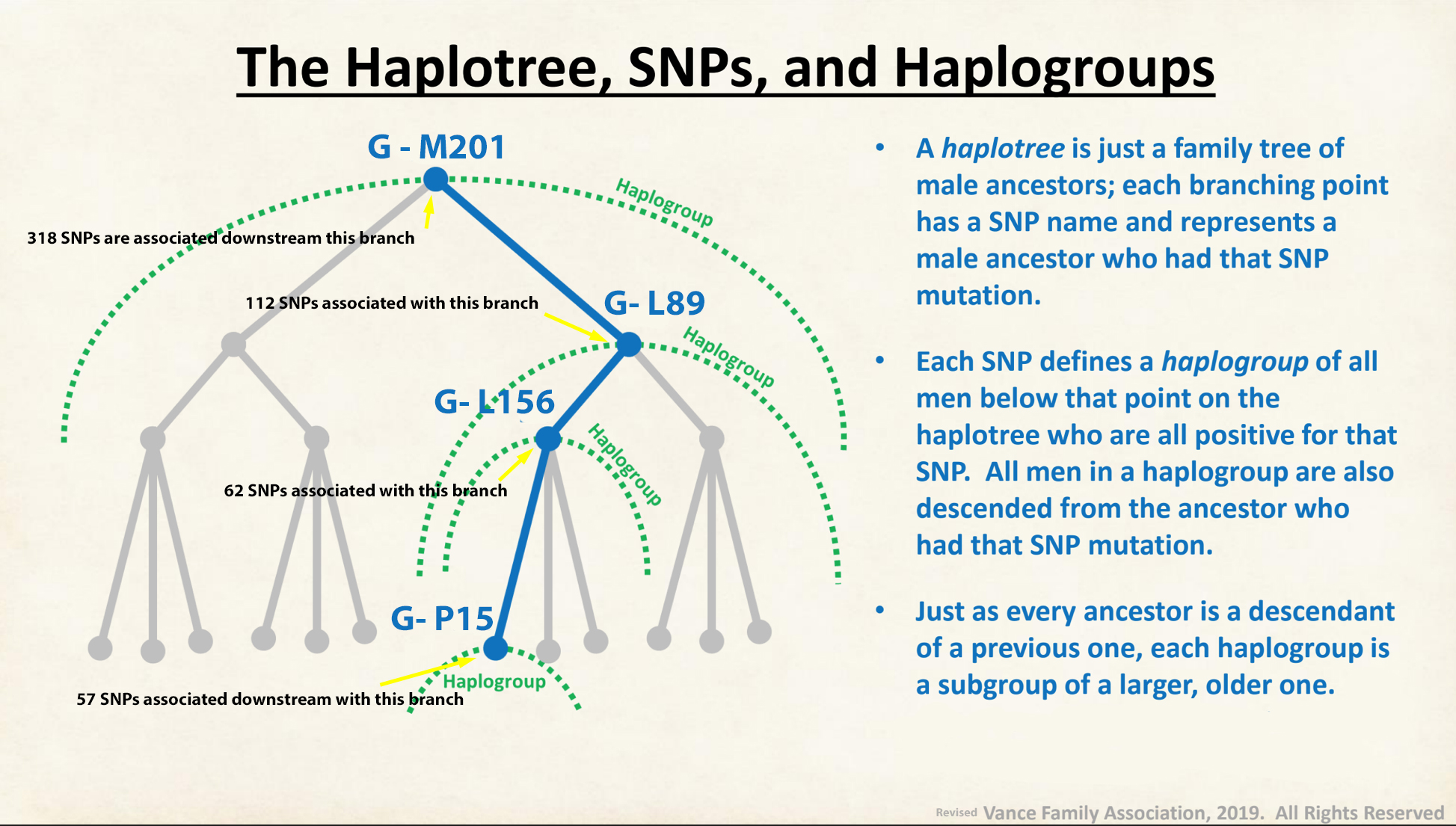
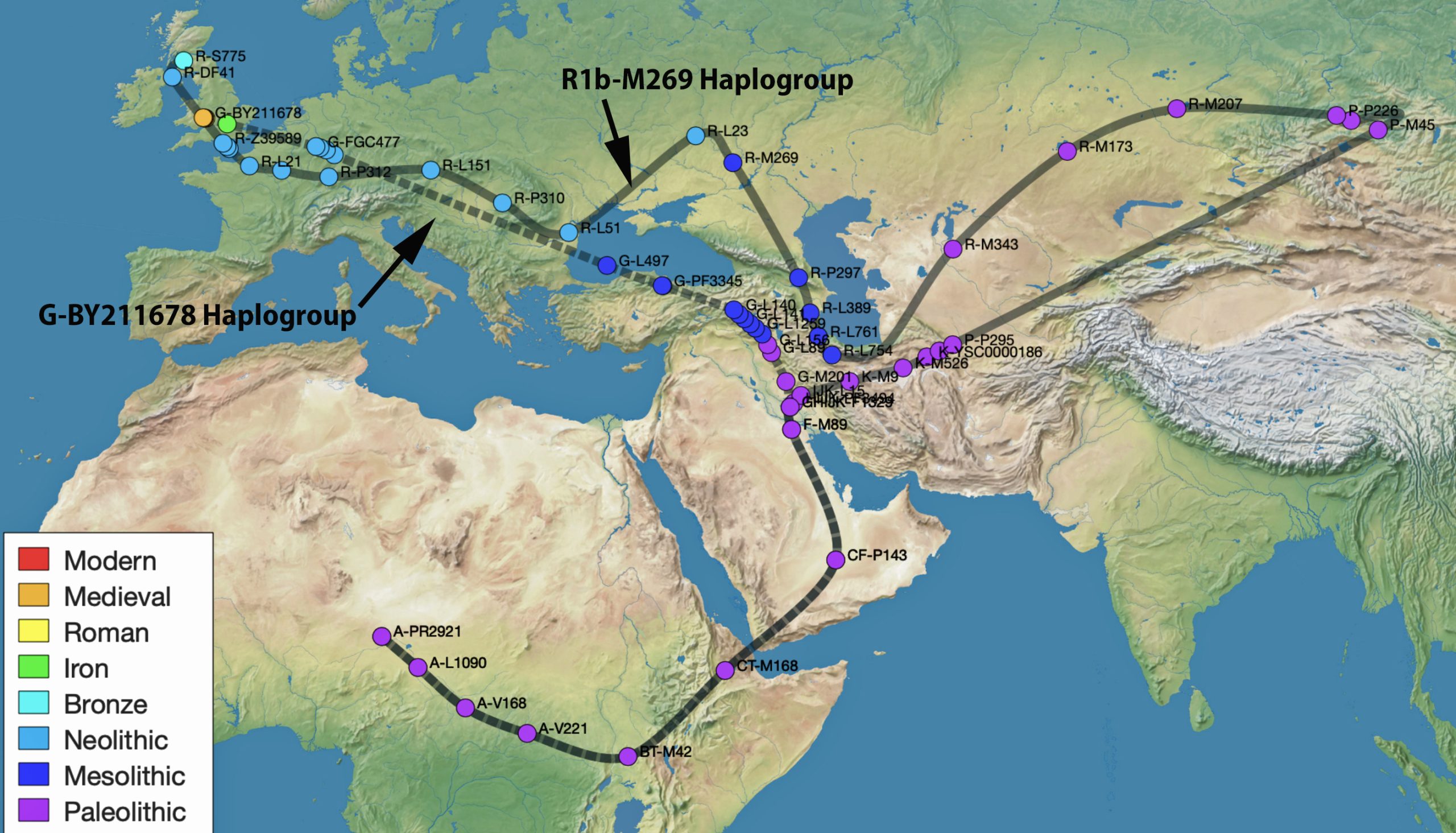
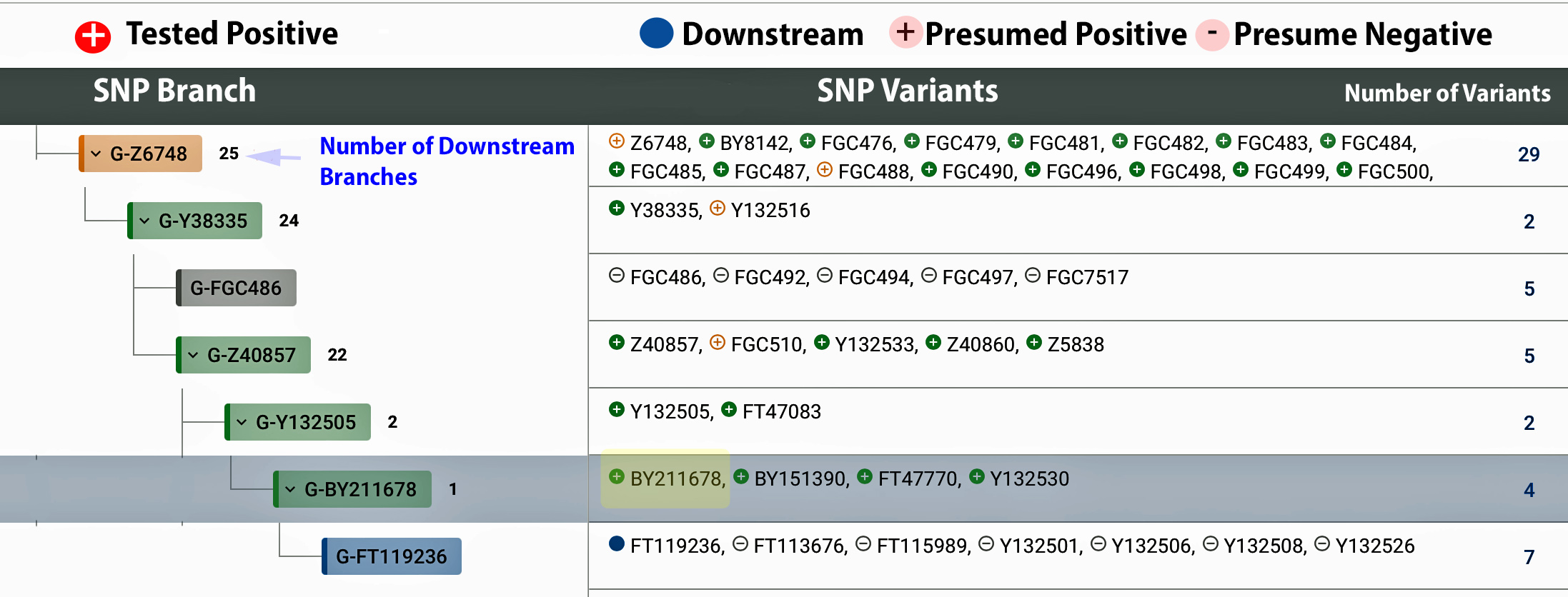
The information in Table One was introduced in Part Three of this story and will be used as a basis for discussing my path of discovery for genetic ancestors using the concept of genetic distance and tMRCA. The table displays Y-Chromosome DNA (Y-DNA) STR results for testers in the L-497 Haplogroup project. As reflected in Illustration One, twelve test kits were grouped together based on how they tested for specific SNPs associated with branches in the haplotree.
Illustration One: The One Two Punch of SNP then STR Analysis
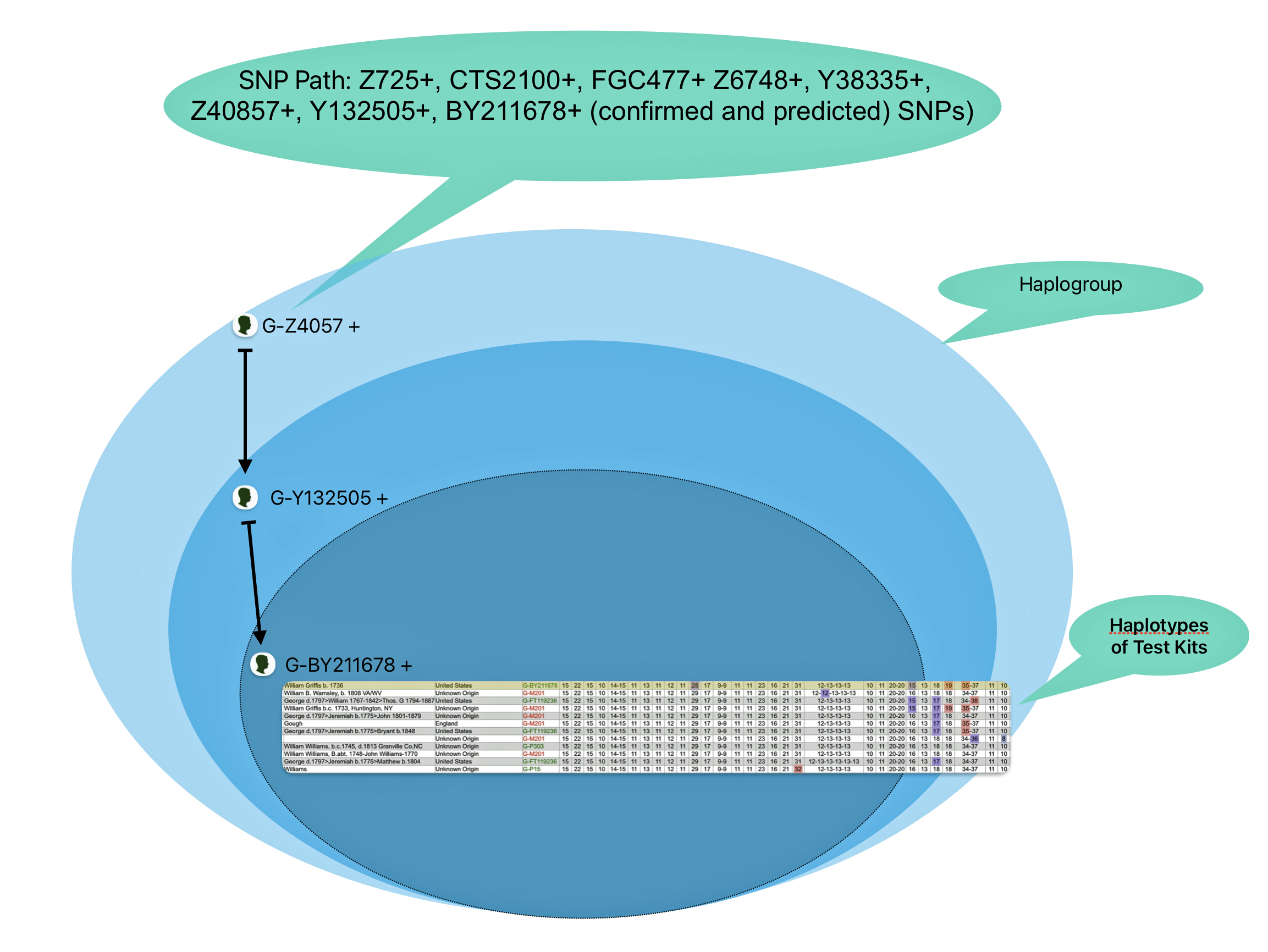
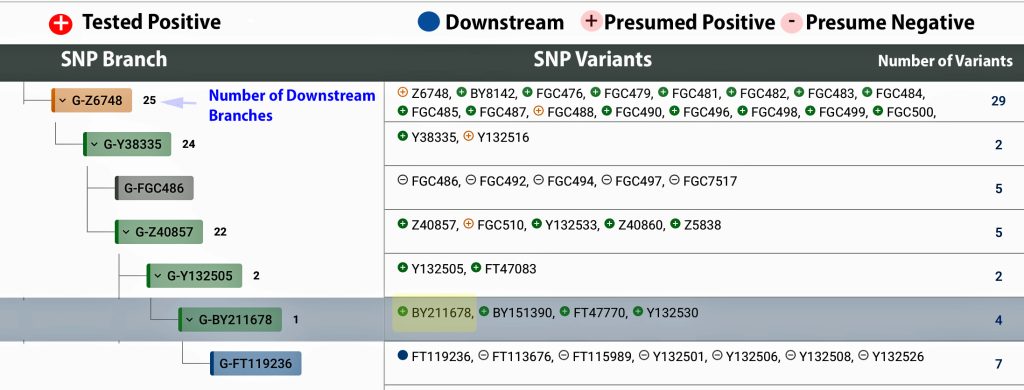
Specifically, Table One provides STR data on my haplotype (STR signature), which is highlighted in the table, for 111 sampled STR values. My results are grouped with eleven other men based on our similarity in our respective STR haplotype signatures. We also share similarities in SNP tests and have been grouped in the G-BY211678 haplogroup.
Table One: 111 STR Results for G-L497 Working Group Members within the G-BY211678 Haplotree Branch

The table provides the modal haplotype for the twelve individuals (re: third row) and the minimum and maximum values for each of the STRs listed in the table. FTDNA uses the concept of genetic distance (GD) to compare and evaluate genetic resemblance of two or more STR haplotypes. It is at this point we start to compare STRs among potential test kits.
Genetic Distance: What Does It Mean, How is it Used & How to Portray It
A haplotype (haploid genotype) is a group of alleles in an organism that are inherited together from a single parent. [8]
Unlike other chromosomes, Y chromosomes generally do not come in pairs. Every human male (excepting those with XYY syndrome) has only one copy of that chromosome. This means that there is not any chance variation of which copy is inherited, and also (for most of the chromosome) not any shuffling between copies by recombination. Unlike autosomal haplotypes, there is effectively not any randomization of the Y-chromosome haplotype between generations. A human male should largely share the same Y chromosome as his father, give or take a few mutations; thus Y chromosomes tend to pass largely intact from father to son, with a small but accumulating number of mutations that can serve to differentiate male lineages.
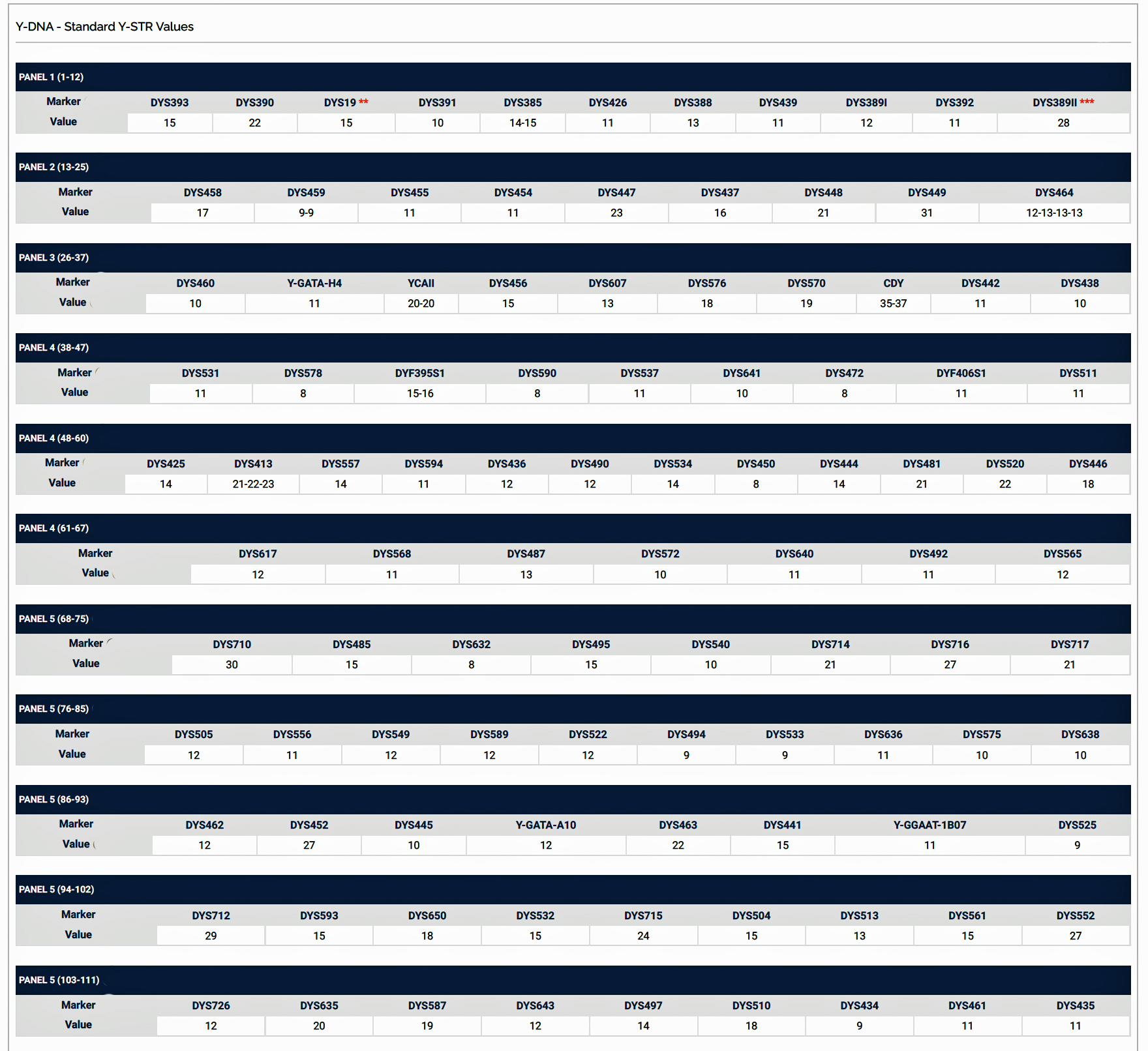
Haplotypes in Y-DNA testing typically compare the results of Y-25, Y37, Y-67, or Y-111 STR tests. Table Two is an example of my haplotype for the Y-111 test. The haplotype basically represents the unique string of values for each of the STRs that compose the test. They number essentially do not mean much by themselves. They take on meaning when you compare them with other testers or pool my results with others to concoct dendrograms and higher level statistical analyses.
Table Two: Example of the Y-111 Haplotype for James Griffis
A modal haplotype is an ancestral haplotype derived from the DNA test results of a specific group of people, using genetic genealogy. Within each FTDNA work group that is based on haplogroups, surnames, geographical area, or other categories, typically test results are grouped on the basis of the most recent common ancestor that is based on a modal haplogroup. [9]
The modal haplotype is found on the third row of the table One. My results are found on the fourth row of the table for Kit number 851614. Click on the image for a viewable version. The table also provides the minimal allele values for each STR marker and the maximum allele values for each marker for comparison.
The ancestral haplotype is the haplotype of a most recent common ancestor (tMRCA) deduced by comparing descendants’ haplotypes and eliminating mutations. A minimum of three lines, as distantly related as possible, is recommended for deducing the ancestral haplotype. This process is known as triangulation. For FTDNA testing, ancestral haplotype basically refers to the haplotype of the tMost Recent Common Ancestor (tMRCA). In genetic genealogy, the Most Recent Common Ancestor (tMRCA) is the ancestor shared most recently between two individuals. [10]
For Y-DNA, the Most Recent Common Ancestor (tMRCA) is defined as the closest direct paternal ancestor that two males have in common . One of the questions all genealogists seek to answer is when a mutation occurs. We want to know when a mutation occurs and how closely we are related to others that have similar SNP or STR mutations. Unfortunately, that question, without traditional genealogical ancestral information, is very difficult to answer.
For the past two decades, many researchers have attempted to reliably answer that question. The key word here is ‘reliably’. The general consensus is that the occurrence of a SNP is someplace, on average, between 80 and roughly 140 years. The topic is hotly debated, and many factors can play into SNP age calculations. [11]
Since STRs mutate faster than SNPs and can also have a likelihood of mutating back to an original configuration, the estimate of the age of a STR mutation is challenging and depends on the specific STR since they each mutate at different rates. Given the nature STRs, the strategy for locating tMCRA with STRs relies on the concept of genetic generations (e.g. genetic distance). Translating genetic distance to years relies on statistical probabilities based on (a) the specific STR markers tested and (2) the number of STR markers used in calculations.
FTDNA Genetic Distance and Y-DNA STRs: Individual Matches
The main feature of FamilyTreeDNA’s Y-STR tests (Y-37 through Y-111) are finding Y-DNA matches. Like most DNA tests for genealogy, the test is most useful when compared to other people. The key question is, “When was the last common ancestor with this match?” When that is not obvious from comparing known genealogies, the genetic distance is the metric used to compare and estimate how far back in time the connection goes to identity the Most Recent Common Ancestor (tMRCA). Is the connection in recent times, just behind that genealogical brick wall, or in ancient, prehistoric times?
The FTDNATiP™ Report (TiP for Time Predictor) translates the Genetic Distance (GD) statistic into a time unit in predicted ‘years ago’. Depending on the average rate of mutation for sampled marker STRs, the number of differences between two samples (individuals) grows larger as the number of generations back to a common ancestor increases. FTDNA uses this idea to limit the number of matches shown in their match reports. As reflected in Table Three, if you have a 12 marker test (Y-12 STR test), their cut off is a genetic distance of one (one mutation difference), for their Y-37 marker tests the report cut off is at a genetical distance of 4, at 67 markers it is 7, and at 111 markers the report cut off is 10. [12]
Table Three: FTDNA Limits on Genetic Distance Based on Level of STR Test
| Test Level | GD Limit for Matches |
|---|---|
| Y-12 | 0 or 1 if they are in the same working group project |
| Y-25 | 2 |
| Y-37 | 4 |
| Y-67 | 7 |
| Y-111 | 10 |
In general, the closer the match in haplotypes between two individuals, the shorter the time back to a most recent common ancestor. For instance, if two individuals share the allele values for 35 out of 37 STR markers, they almost certainly share a more recent common ancestor than two individuals who share 25 out of 37 markers.
When it comes to calculating the genetic distance of a common ancestor, which STRs are different between the two individuals is more important that how many differences there are. This is due to the fact that STRs can behave differently from their expected mutation rates and because some STRs mutate faster than others. Regardless of whether one takes a 12 37, or 111 STR marker test, a distance of four matters more based on the mutation rates for each of the four markers that are different.
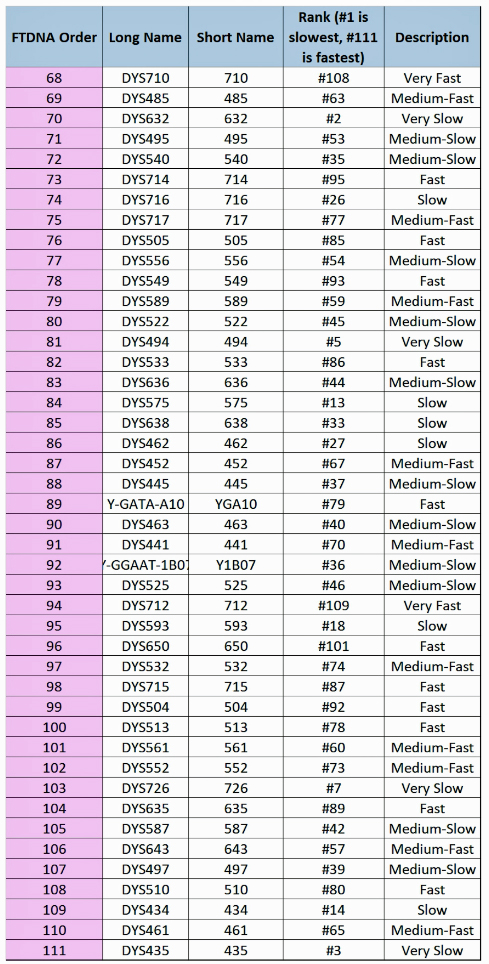
The following tables indicate the mutation rates for each of the STRs that are used for the various STR tests. [13]
Table Four: Mutation Rates for STRs 1 Through 37
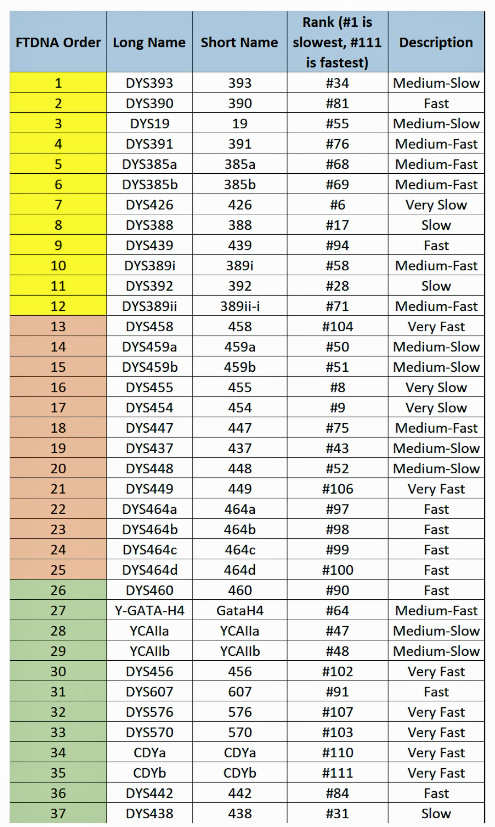
Table Five: Mutation Rates for STRs 38 – 67
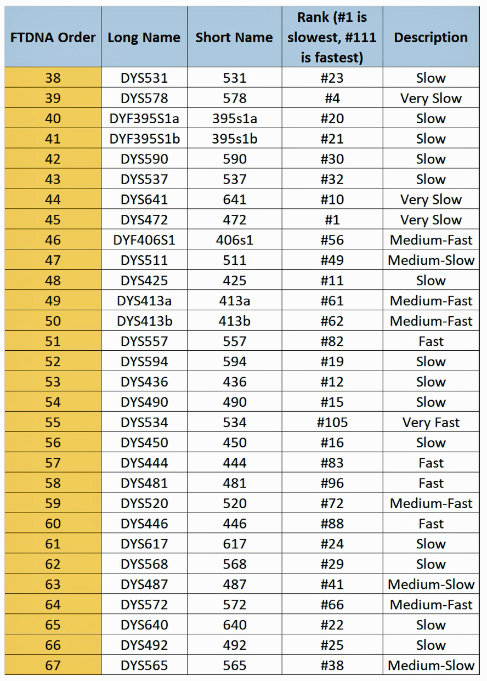
Table Six: Mutation Rates for STRs 68 – 111
As mentioned earlier, calculating the Time to Most Recent Common Ancestor is based on probability and is not an exact science. We can identify the most likely time that a common ancestor might have lived, but there will always be a degree of uncertainly. It is better to think of “the Most Recent Common Ancestor” (tMRCA) as a range of time rather than a point in time. [14]
The following four charts show (noted by the dark line) the average number of generations that Y-DNA matches will share a common ancestor based on genetic distance. The statistical confidence levels are based large population samples and the two lighter lines show a band or range in which 95 percent of the matches will fall. The charts indicate where the FTDNA ‘cut off’ occurs. Notice that as you test more STR markers, the genetic distances also go up for the same number generations. For the Y chromosome these rates assume a 31 year generation and basing years ago from a 1955 “present date”. [15]
As illustrated in the following four illustrations, the statistical variabiability in determining the range of generations based on the concept of genetic distance can vary widely. Even comparing genetic distance with 111 STR test results, one will have a wide statistical variance. A genetic distance of 2 for a Y-111 comparison will mean that the match is within a 95 percent confidence interval of 2-10 generations. If a generation is around 31 years, then the match is equivalent to 62 – 320 years. Translating this range to ‘years before present would be 1955-62= 1893 CE and 1955-320= 1635 CE. That can be a wide range if you are looking for genetical matches. [16]
Illustration Two: Relationship of Genetic Distance to Generations at Y 12
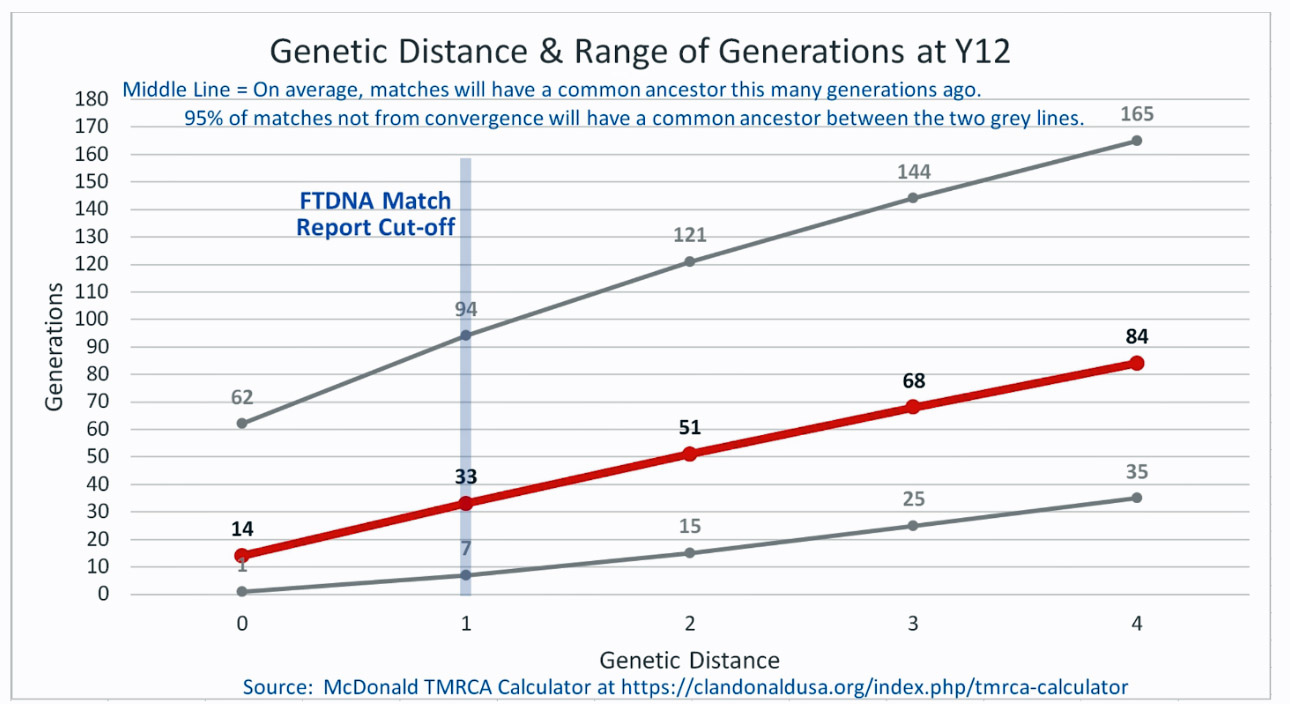
Illustration Three: Relationship of Genetic Distance to Generations at Y37
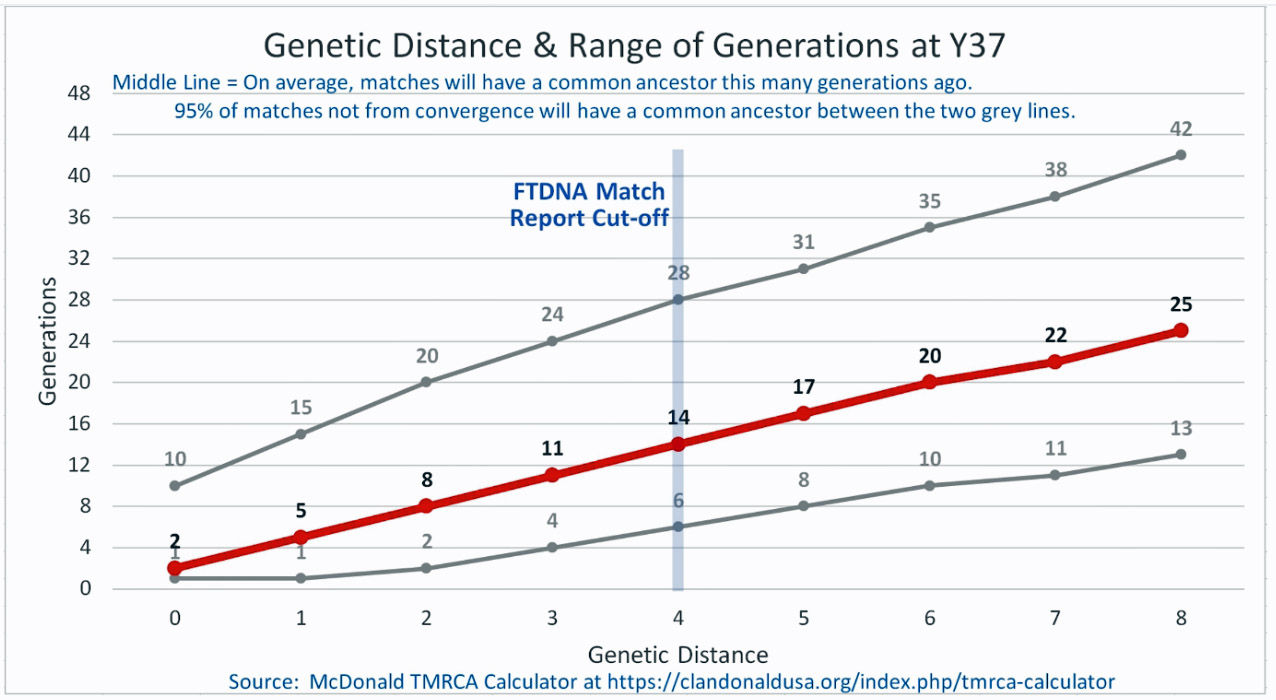
Illustration Four: Relationship of Genetic Distance to Generations at Y67
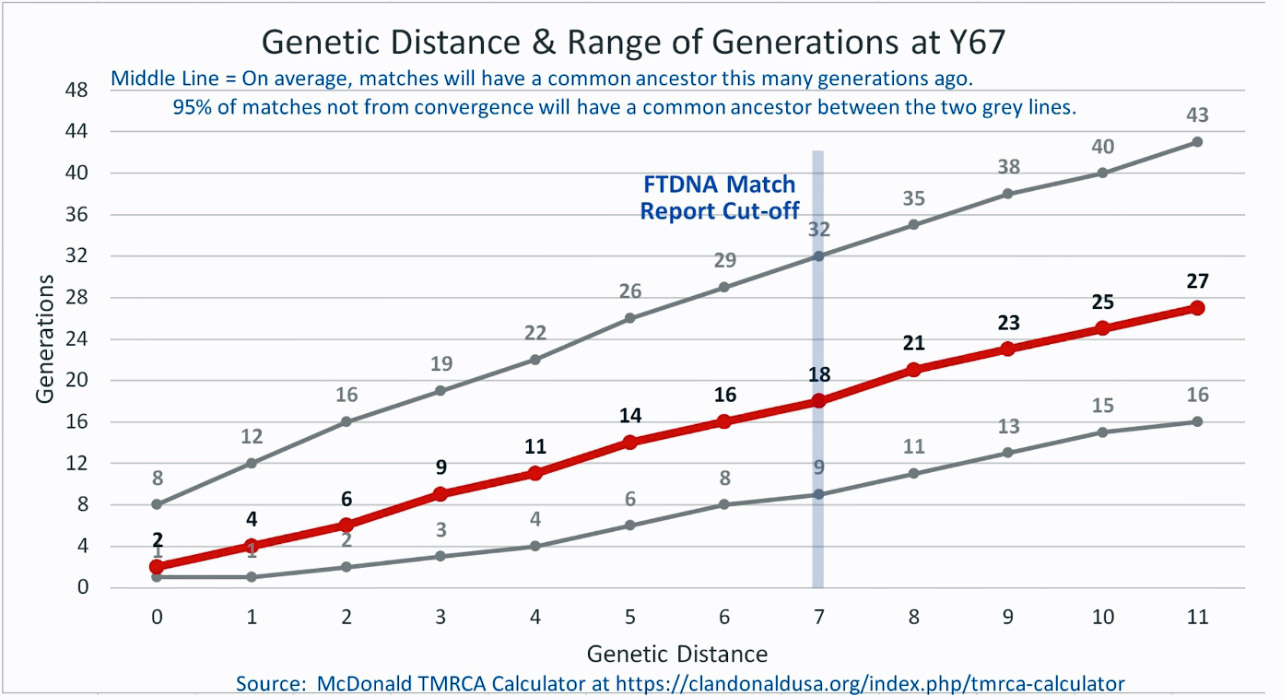
Illustration Five: Relationship of Genetic Distance to Generations at Y111
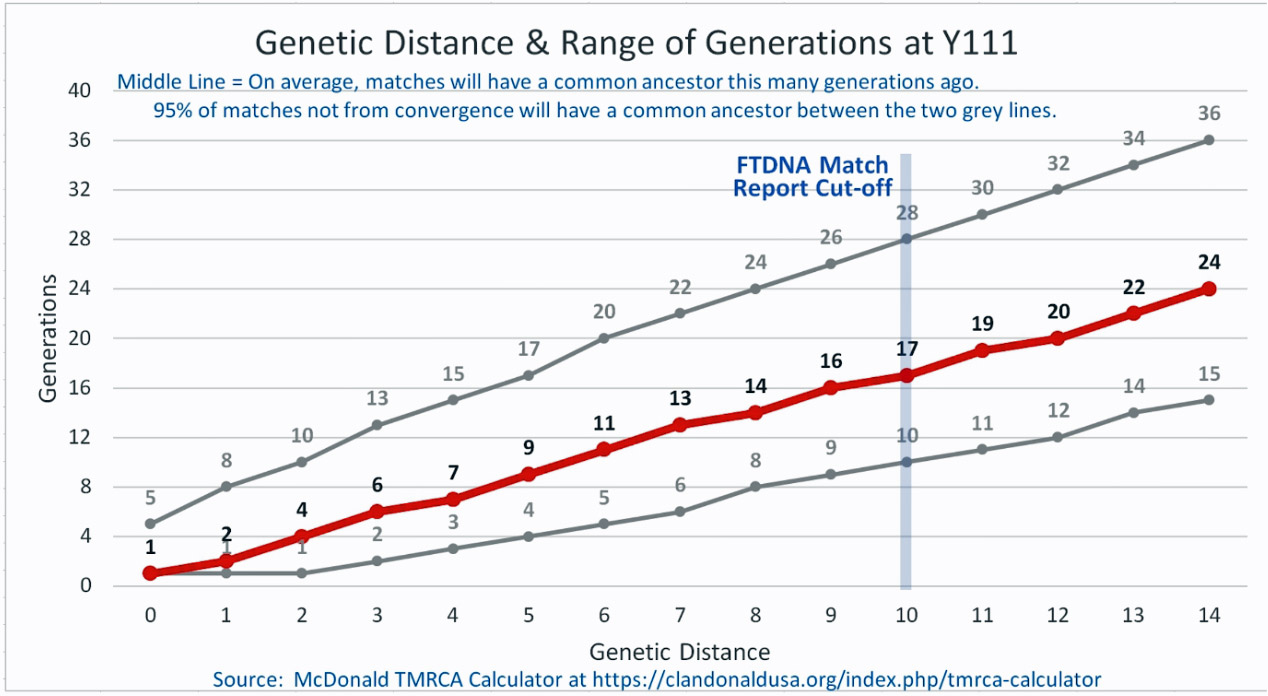
Up until very recently, there were two methods to determine the GD.: the Step-Wise Mutation Model and the Infinite Allele Model. [17] In 2022, FTDNA released Age Estimates based on the Big Y-700 test. test results The millions of slow-mutating Y-SNP markers tested by Big Y together with the faster-mutating but fewer Y-STR markers derived revised the Time to Most Recent Common Ancestor (TMRCA) estimates of each branch on the Y-DNA haplotree. [18] Also in 2022, FTDNA updated FTDNATiP™ Report using Big Y haplotree TMRCA estimates from hundreds of thousands of pairs of Y-STR results from Big Y testers and built models to predict the most likely TMRCA ranges for each Y-STR marker level and genetic distance. [19]
Most mutations only cause a single repeat within a STR marker to be added or lost. For these markers, the Step-Wise Mutation Model is used. For example in Table Seven, comparing my results (Kit Number 851614) with Kit number 125476, who also lists a William Griffis as a Paternal Ancestor, the values of two STR markers differed by one value (see below), which means our GD is 2.
Table Seven: Comparison of Two STR Markers
| Kit Number | DYS389ii Allele Value | DYS576 Allele Value |
|---|---|---|
| 851614 | 28 | 18 |
| 125476 | 29 | 17 |
In some cases, an entire marker is added or deleted instead of a single repeat within a marker. This is believed to represent a single mutation in the same way that the addition or subtraction of a repeat is a single mutation event. For this reason, FTDNA uses the Infinite Allele Model in these cases. When an STR simply does not exist in an individual, this is called a null value. When a marker is missing, the value is listed as 0.
Multi-copy STR markers appear in more than one place on the Y chromosome. These are reported as the value found at each location, separated by hyphens. For example, in table one you may see DYS464= 12-13-13-13 or 12-12-13-13-13 or 12-13-13-13-13-13 . This means that the STR marker DYS464 has a unique number of repeats in each location. These locations are usually referred to as DYS464a, DYS464b, DYS464c, etc.
An example of this situation is illustrated in Table Eight by comparing my STR results in Table One (Kit Number 851614) with Kit Number 31454 (whose Paternal Ancestor is William Wamsley) and 285488 (whose self reported paternal ancestor was George Williams).:
Table Eight: Comparison of Multi-Copy STR Markers
| Kit Number | DYS 464a | DYS 464b | DYS 464c | DYS 464d | DYS 464e | DYS 464f | Total GD |
|---|---|---|---|---|---|---|---|
| 851614 | 12 | 13 | 13 | 13 | |||
| 31454 | 12 | 12 | 13 | 13 | 13 | 2 | |
| 285488 | 12 | 13 | 13 | 13 | 13 | 13 | 2 |
Within multi-copy markers, there are two types of mutations, or changes, that can occur: marker changes and copy changes. Marker changes (changes in how many repeats are within a marker) are counted with the Step-Wise Mutation Model. Copy changes (changes in the number of markers, regardless of how many repeats are in each) are counted with the Infinite Allele Model.
In the example illustrated in Table Eight, if we compare Kit 31454 to my kit 851614, the allele value for DYS464b is different by one (marker change) and also 31454 has an additional copy (DYS464e), which totals to a genetic distance of 2. Comparing kit 285488 with my kit reveals no marker changes in DYS464a-d but two additional copy changes (DYS464e-f), which totals to a GD of two.
Adding together the GD for each marker in two people provides the overall GD for those two people. When a GD becomes ‘too great’, it is unlikely that the two people share a common ancestor within a ‘genealogical timeframe’, so FTDNA establishes a upper level limit for reporting matched based on GD.
Table Nine provides a practical example of FTDNA’s strategy of comparing the differences between haplotypes of individual test results based on similar haplogroups. I have listed the surname of each of the testers and the STR test they completed (re: Y-37, Y-67, Y-111, or Big Y 700 test. The table also provides information on the most recent haplogroup branch their respective tests were able to document. A Big Y 700 test provides results for 700 STR and therefore can provide the most granular test results for haplogroup designation. The table also indicates the self reported earliest known paternal ancestor for the tester.
Table Nine: STR Haplotype Matches with James Griffis Based on Y-37 Test
| Kit No. | Surname | STR Markers Tested | Genetic Distance (GD) | Likely Common Ancestor (Genera- tons) [12] | MRCA Based on GD [12] | Earlest Known Ancester |
|---|---|---|---|---|---|---|
| 125476 | Griffith | 37 | 2 Steps | 8 (2-20) | 1650 CE | William Griffis |
| 39633 | Compton | 37 | 2 Steps | 8 (2-20) | 1650 CE | Unknown |
| 154471 | Williams | 111 | 4 Steps | 3(7-15) | 1700 CE | William Williams |
| 285488 | Williams | 700 | 4 Steps | 3(7-15) | 1700 CE | George Williams |
| 294448 | Williams | 111 | 4 Steps | 3(7-15) | 1700 CE | George Williams |
| 285458 | Williams | 111 | 4 Steps | 3(7-15) | 1700 CE | George Williams |
| 36706 | Williams | 67 | 4 Steps | 11(4-22) | 1500 CE | William Williams |
| 149885 | Gough | 37 | 4 Steps | 14(6-28) | 1300 CE | Gough |
As illustrated in Table Nine, although the tester whose last name is Griffith (first. row of the table) only tested for the Y-37 test, his test results are 2 steps different from my test results. If we look at Illustration Three above, this means I and Mr. Griffith share a common ancestor around 8 generations ago or between 2 to 20 generations.. Eight generations would be around the revolutionary war period.
There is another test kit that is 2 steps different from my test kit results. The test kit 39633, who has a surname of Compton appears to be as close as Mr. Griffith. I do not have any traditional genealogical documentation that references an individual with the last name of Compton. Rather than dismiss the results, one needs to look ‘outside the box’ in terms of critically analyzing the results. I may need to reach out to this gentleman to see what potential connections we might have. Also, based on the statistical confidence levels associated with the Y-32 STR tests, the MRCA may be as far back as 20 generations or 620 years ago which is around 1400 CE.
The remaining six testers are four steps different from my test results. While I know there are no individuals who are related in the past three generations, perhaps 15 to 22 generations back there might be a common ancestor. The outer range would be around 682 years ago or around 1340 CE. which would be before the use of surnames.
Based on the results, further research into the background of Mr. Griffith, whose earliest known ancestor was a ‘William Griffis from Huntington, NY” may lead to promising results! 12 generations would be around the early colonial era (1650). It may also be worthwhile to look into the Williams’ connections!
Phylogenetic Trees: Graphic View of Genetic Distance at the Lineage Level
In addition to analyzing and providing Y-DNA test results, FTDNA provides a wide platform of ways in which DNA results are analyzed and the results are packaged for consumers to identify possible genetic matches. There are also a number of analytical tools that have been developed by individuals that compliment or enhance the ability to assess genetic distance.
I can complement the second stage of an analysis by reviewing the results of genetic distance that we just discussed in a number of program generated mutation history trees. These types of programs give a pictorial representation of how the different members of a lineage may be related.
The branching pattern derived from the DNA mutations may very well correspond to the branching pattern that one might see in a traditional family history tree if we were able to trace it all the way back with documentary evidence to the MRCA (Most Recent Common Ancestor). The Mutation History Tree can give us important clues regarding which individuals are likely to be on the same branch of the overall tree, and who is more closely related to whom. This in turn can help focus further documentary research.
One type of mutation history tree has been developed by David Vance that uses FTDNA data that creates a Y-DNA phylogenetic tree. The program is relatively easy to use and graphically provides an intuitive approach to visualize the possible genetic relationships between various DNA test results. The program is referred to as the SAPP analysis (Still Another Phylogeny Program). The current version that was used in my analysis was SAPP Tree Generator V4.25. [20]
The program uses STRs from any of the STR tests (e.g., Y25, Y37, Y67, Y111), to construct a Y-DNA phylogenetic tree. It also has the ability to incorporate the SNPs found in BigY tests to fine-tune the genetic links and estimated times to the most common recent ancestor. The program can also incorporate known names and birth dates of ancestors to further fine-tune the analysis.
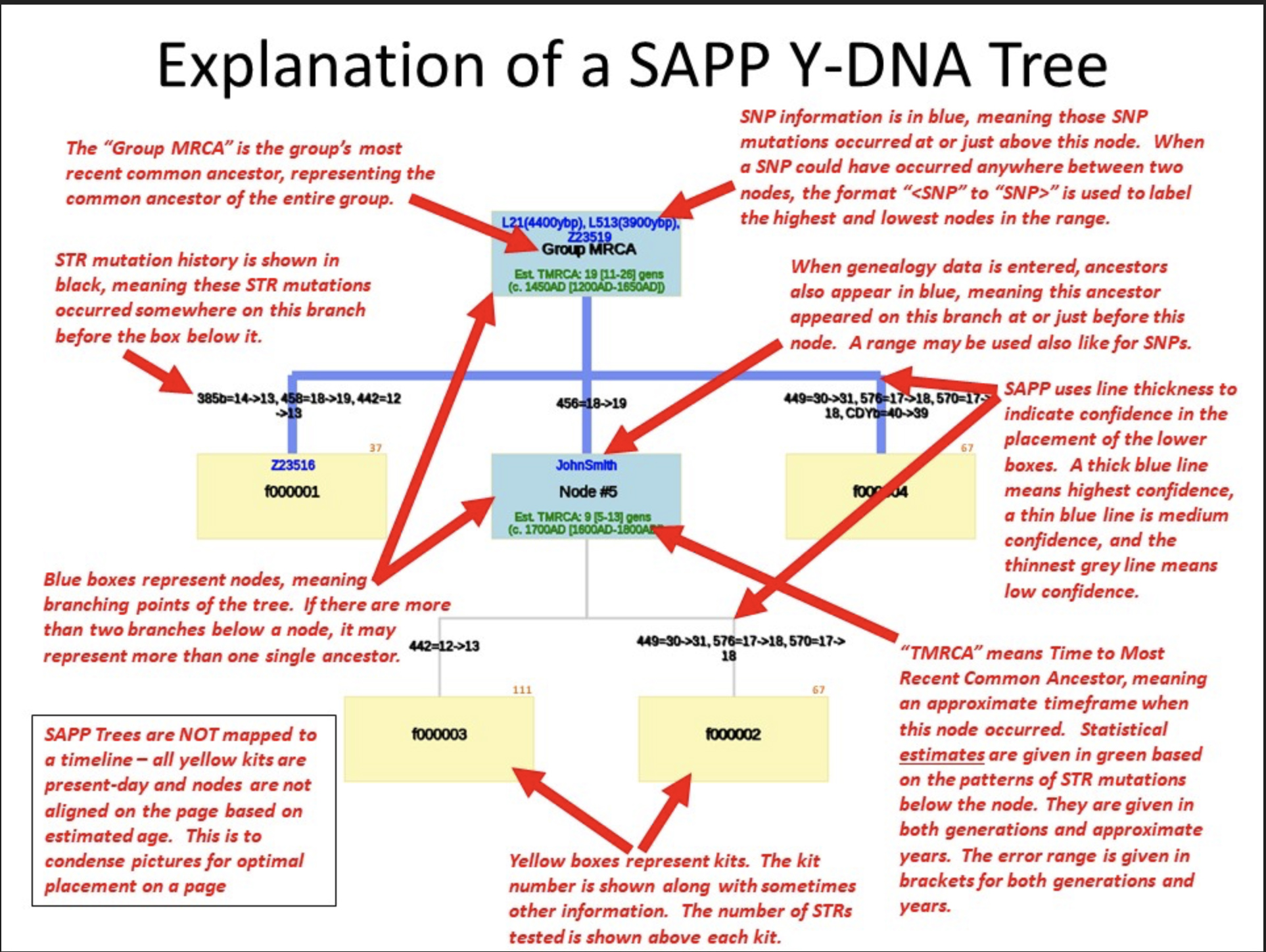
The program provides:
- STR Table. This table is included to verify the STR input. It starts with the calculated Group Modal Haplotype for your input set followed by all the input kits with the off-modals colored.
- Original Genetic Distance Table. This table calculates genetic distances (GDs) from the input STR results. It should match closely with GD calculations from other tools and commercial companies.
- Adjusted Genetic Distance Table. This table re-calculates the GDs based on the tree that SAPP has just calculated. It will correct for any convergence that may have occurred in the calculated tree.
- Kit to SNP/Genealogy Cross-Reference. This table summarizes the input SNP and Genealogy data showing the +. -. or ? status against the various kits.
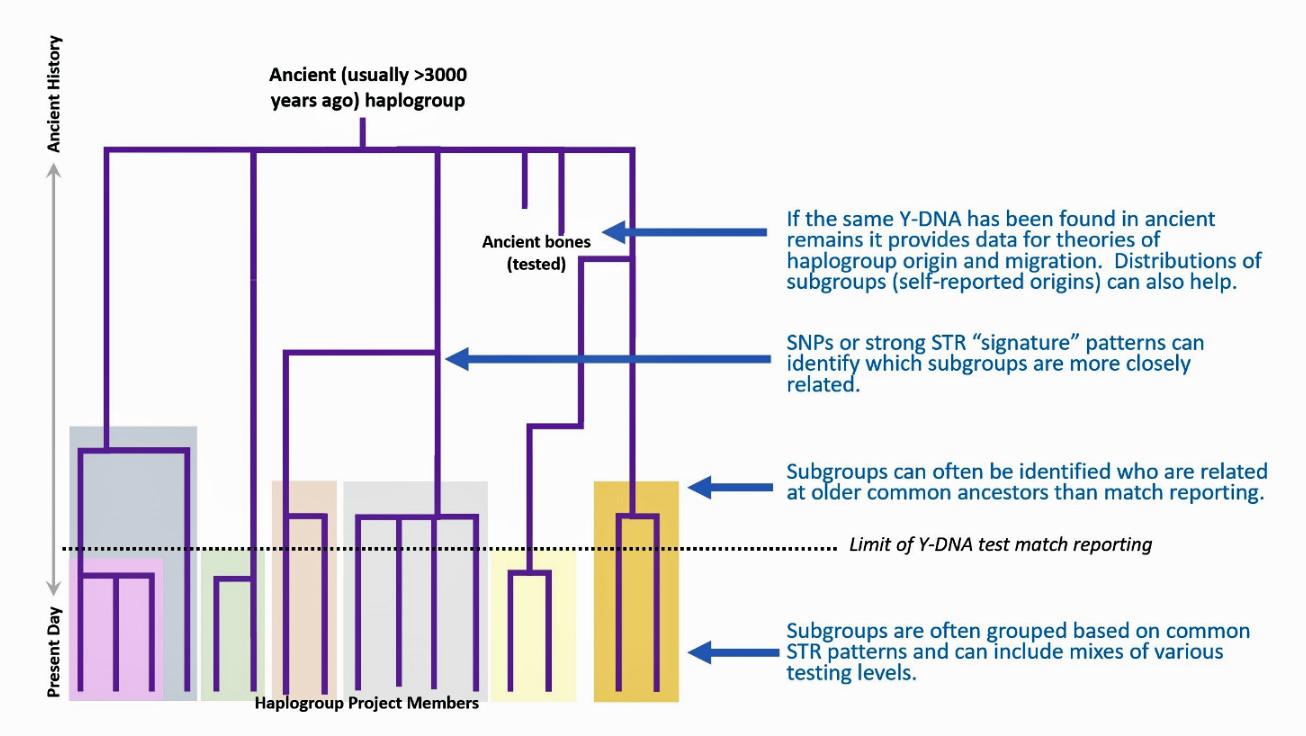
- The Image or Web version of the Tree File. The program creates a downloadable file containing the phylogenetic tree. Normally the tree is drawn as a graphic, as indicated in Illustration Six.
Illustration Six: Explanation of the SAPP Phylogenetic Tree
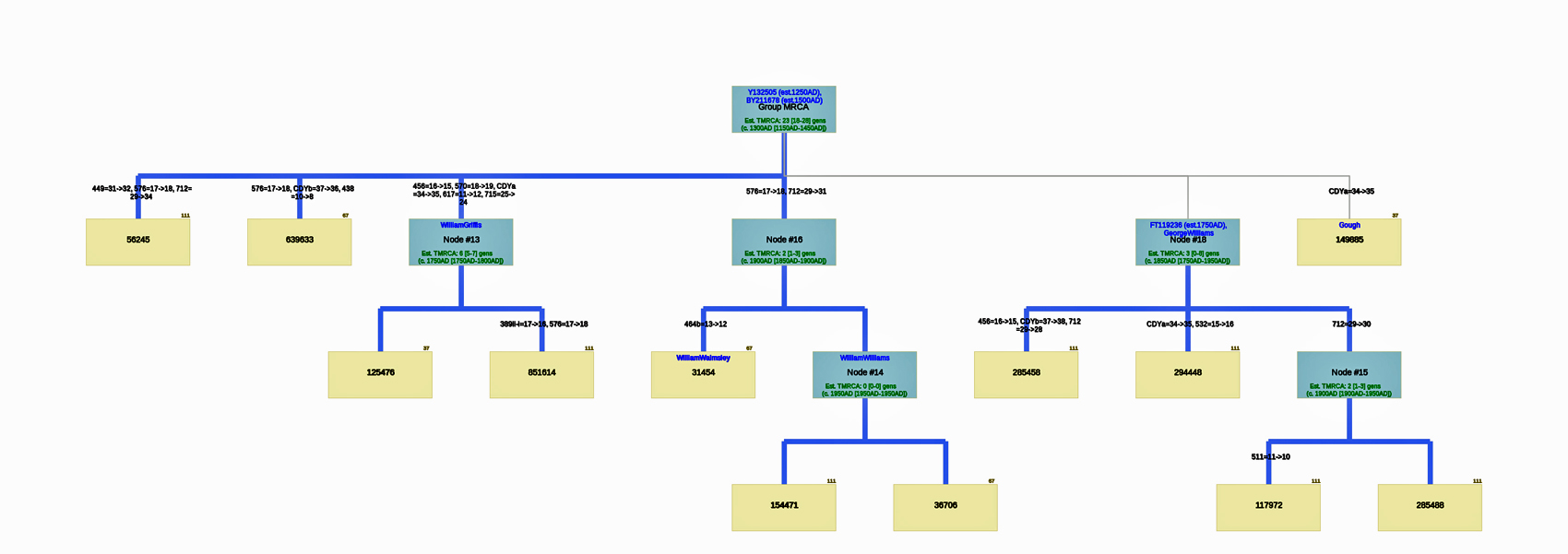
Utilizing the STR results, SNP data, and self reported paternal ancestor information for the 12 tests kits found in Table One, the following phylogenetic tree was created (click on the image of the thumbnail of the tree to be able to actually see the table). I have provided a PDF version of the Phylogenetic Chart which allows you to enlarge the image to get a better view.
Illustration Seven: Phylogenetic Tree Results for FTDNA STR Test Results for Individuals within the G-BY211678 Haplogroup (Click for Larger View)
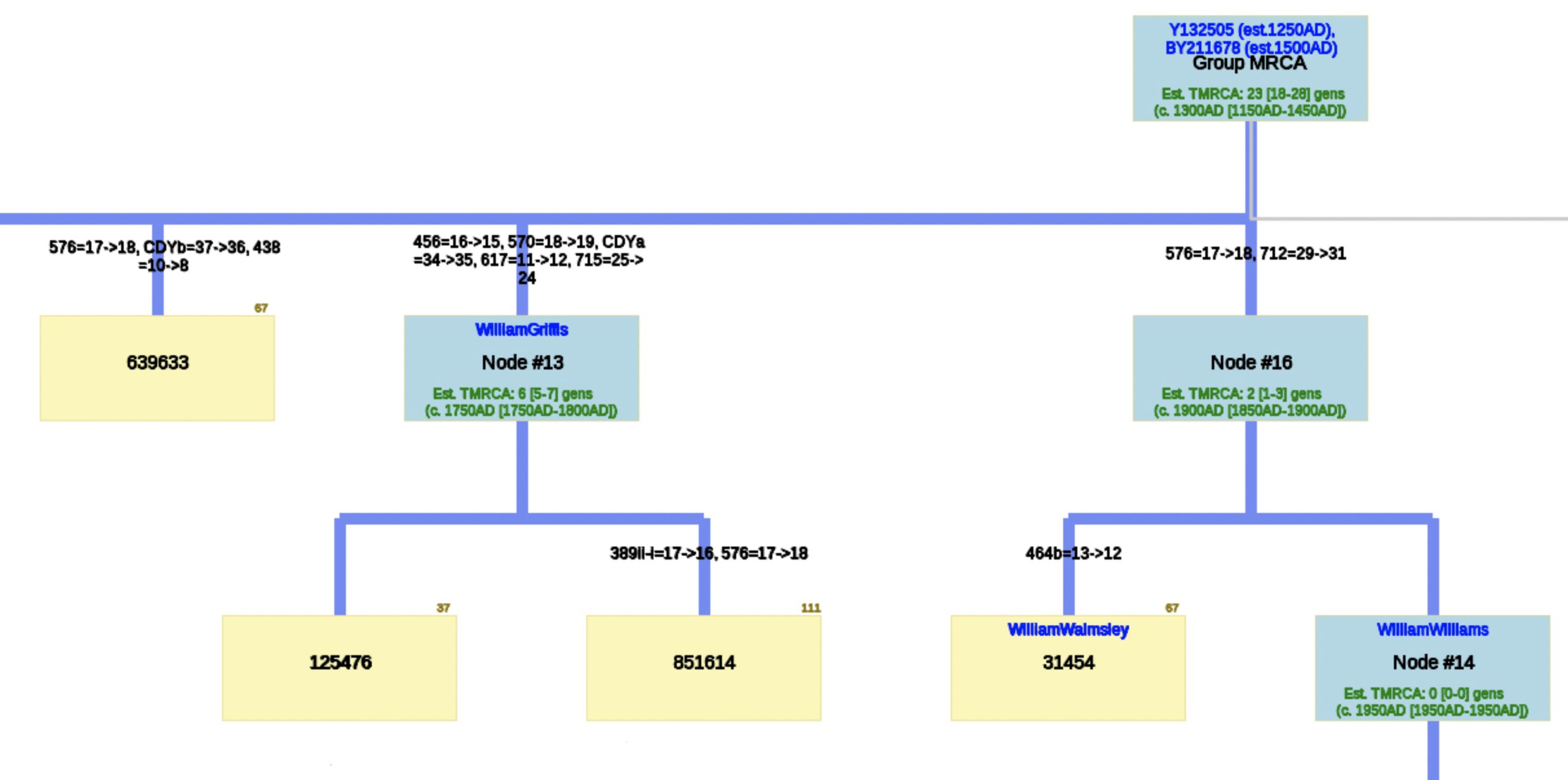
The phylogenetic tree reveals three major genetic groupings of the 12 test kit results. One of those groupings tie my results (FTDNA Kit Number 851614) with an individual whose surname is Griffith (FTDNA Kit Number 285458) and claims the same paternal ancestor, William Griffis see Illustration Eight.
Illustration Eight: Close Up of Phylogenetic Tree
The following are the original and adjusted genetic distance tables generated by the SAPP program. The number of STRs tested are listed on the diagonal in blue. Cell colors refer to the number of STRs tested – cells of different colors are not directly comparable.
Red numbers indicate where adjusted genetic distances are different from original calculation.
Table Ten: SAPP Generated Original Genetic Distance between the 12 Test Kits.
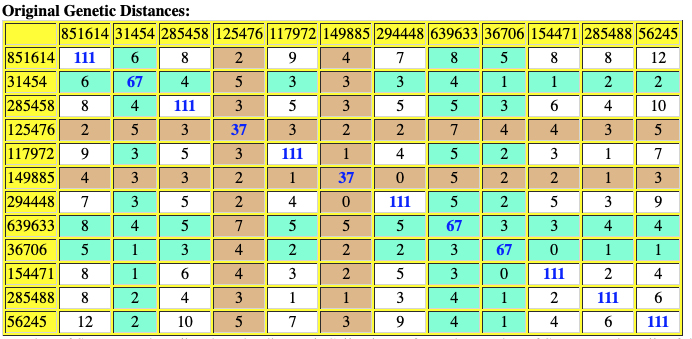
Table Eleven : SAPP Generated Original Genetic Distance between the 12 Test Kits.
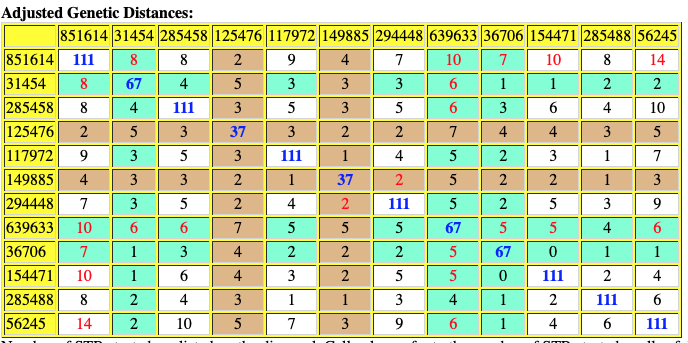
Based on the SAPP results, consistent with the FTDNA analysis, it is estimated that the most recent common ancestor between me and Mr. Griffith is approximately 8 generations or 248 years ago (estimating a generation to be 31 years) which would mean the MRCA was born around 1772. The birth date of William Griffis was 1736.
The results of the SAPP analysis suggests that there possibly may be an additional three haplotree branches, based on differences between STR haplotypes among the twelve test kits.
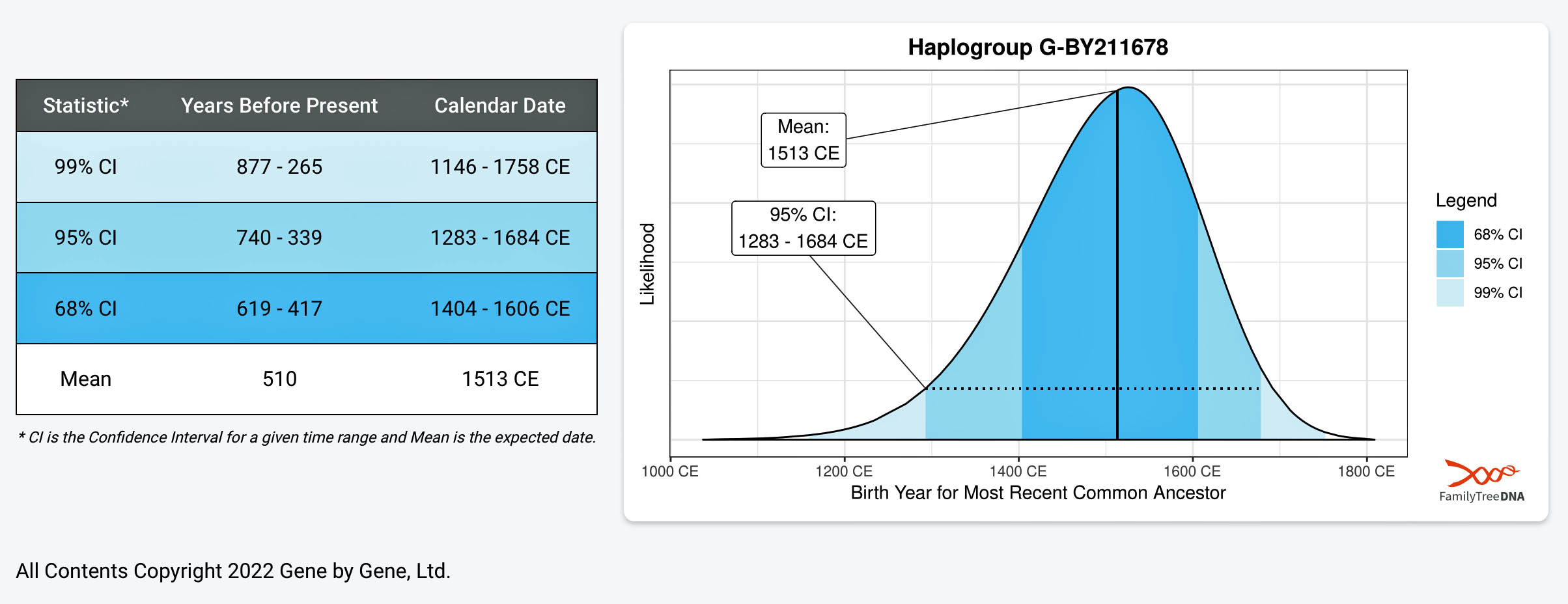
The phylogenetic chart indicates that the MRCA for all of the twelve test kits is estimated at 23 generations. The MRCA was born around 1500 CE for the G-BY211678 haplogroup. The Node #13 of which I and Mr. Griffith are representatives has the strongest connection in the tree. M=Test kits that indicates the ancestral person as William Williams or William Walmsley appear to have a MRCA 3 generations ago (born around 1850).
Genetic Distance at the Macro Level: Distance Dendrograms
The creation of dendrogram is another tool to use when analyzing STR data. Dendrograms can provide insights into macroscopic patterns in Y-DNA genetics and possible genetic matches of present day Y-DNA testers. The diagram based approach of a dendrogram is visually intriguing. Distance dendrograms are software-generated diagrams that convey relationships based on distance measured either in years or generations. Statistically, the dendrograpms used in the present context for genealogy are constructed by hierarchic clustering and the UPGMA method and are more focused on macroscopic genetic patterns. They complement other tools that focus on family level matches. [21]
Up until this point in the story we have discussed computing tMRCA based on the concept of genetic distance (GD). This sort of pairwise tMRCA analysis is subject to a signfiicant range of statistical uncertainty (as reflected in the above tables for generational distance).
A tMRCA can also be calculated between a single DNA tester and the estimate pattern of a chosen ancestor using a modal haplotype. If you have a large enough set of DNA test kits to sample, the ancestral haplotype will be close to that unknowable MRCA. However, this type of averaging still creates a wide level of variance for individual contemporary testers to compare their results with this ‘statistical archetype’.
The dendrograms generated in Rob Spencer’s model is based on a ‘whole-clade’ estimation of the MRCA. The MRCA for an entire clade (haplogroup branch) can be determined based on a common ancestor or a target SNP. The distribution of pairwise MRCA’s for a number of selected DNA kits in a given clade can be fit into a statistical curve fitting process (e.g. lognormal distribution). This curve fitting process is done on a specific group of DNA kits using statistical methods that are way above my pay grade. [22]
The scale of the data and graphics can reveal large scale, high-level patterns of when one person became the descendant of all others (single founder clades), patterns of descent from a single colonial founder in the America (typically one person is the descendant of all in America), and other demographic patterns that are not apparent using other methods of presenting DNA test results.
Dendrograms are ‘close cousins’ to family trees. The Y-STR Dendrogram is a diagram similar to a family tree. Individual DNA testers are the dots at the right (if the dendrogram is horizontal) or at the bottom (if the Dendrogram is vertical). Time moves backward to the left (if horizontally depicted) or down and up( vertically presented). On a traditional family tree, branch points are actual ancestors. In the dendrogram the branch points are generally not specific people but points in time when genetic mutations or changes occurred. In some cases, with good paper genealogy, branch points can be matched to specific ancestors. [23]
Looking at dendrograpm from another angle, they are graphic renderings of a statistical analysis which compares the differences of STR allele values between a group of DNA testers to determine the most recent common ancestors (tMRCA) between a group of testers. One of the key properties of a distance dendrogram is that if the input distances are accurate and consistent, then the graphic will completely and correctly represent a family tree. If we had a sufficient set of testers who had done DNA tests and tMRCAs could be calculated for all pairs with complete accuracy, then the dendrogram would be an accurate family tree.
You can demonstrate the relatiohsip between dendrograms and family trees for yourself with the Distance Tree Introduction interactive tool, and also for larger and more realistic family trees with the Family Simulator, both created by Rob Spencer.
The major limitation to the accuracy of the dendrogram trees is the statistical and random nature of STR mutations. In general, dendrograms constructed from Y12 or Y37 data will be reliable, while those built with Y111 or Big Y700 data data will be sufficient to see large-scale patterns (“macro genetics”) and in many cases can be close approximations to the true family tree. [24]
One important difference between a dendrogram and a family tree is that a dendrogram defines only the “leaf nodes”. A dendrogram does not “know” that there are other nodes that represent people on the diagram. The joining nodes or points are mathematical constructs. Every joining-point or “T” junction in the diagram corresponds to a specific genetic ancestor.
“(Dendograms) are very reliable for exclusion: you can say with very high confidence that two people are not related if there is a strong mismatch of their STR patterns. This is the forensic use of DNA: it’s very powerful in proving innocence while less decisive about proving guilt.” [25]
“Most of us use Y STR data locally to explore personal matches and to help in building family trees. But STRs can tell us much more when we sit back and take a long look. In this talk we use an efficient way to visualize thousands of kits at once. The large-scale patterns explain “convergence”, illuminate ancient, feudal, and colonial expansions, pick apart Scottish clans, identify American immigrant families, allow accurate relative clade dating, let us see the onset of surnames, and reveal the power law distribution of lineages.” [26]
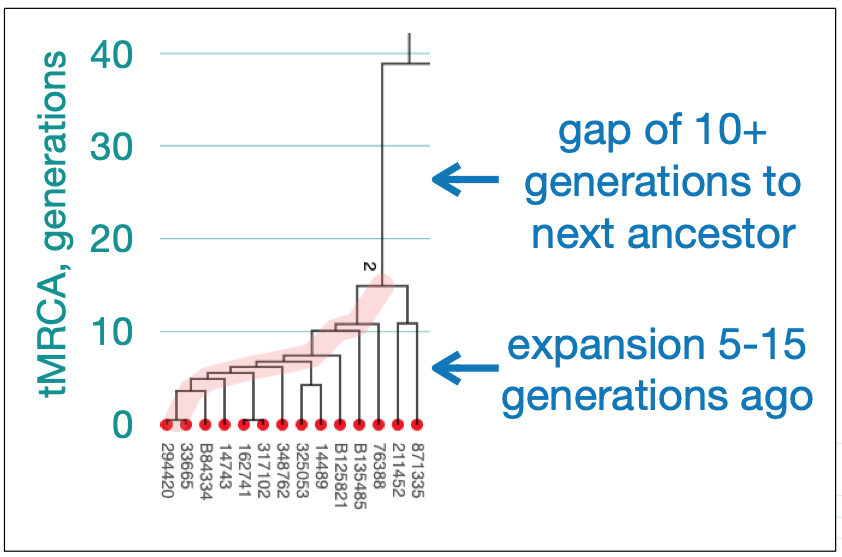
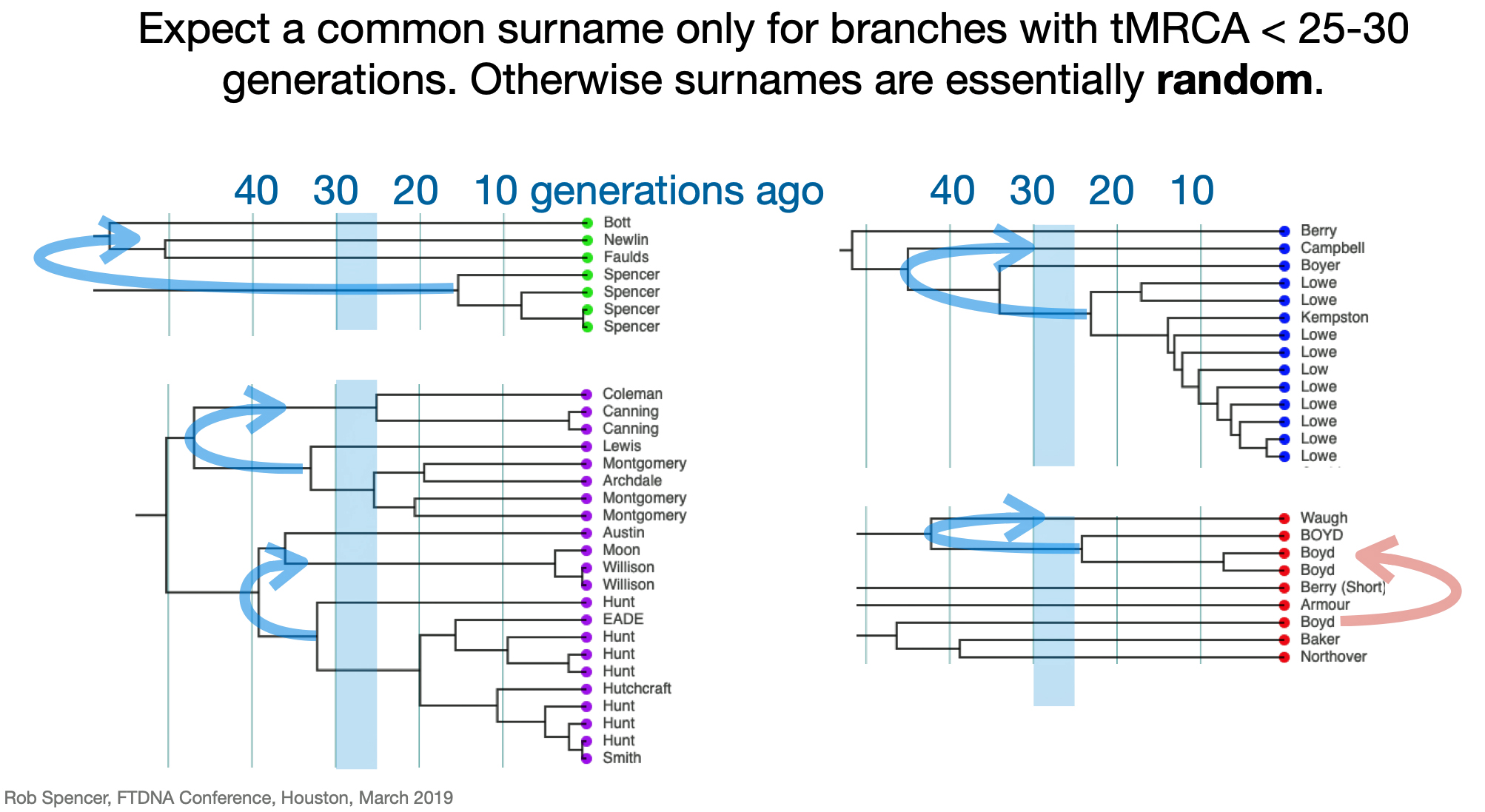
Utilizing STR and SNP data, dendrograms can spot American Immigrant families based on the shape of the dendrogram. Typically there is a gap of 10 plus generations to the next ancestor and an expansion around 5-15 generations ago. [27] Similarly, the advent of surname usage can appear in dendrogram renditions of Y-DNA data. You should expect a common surname only for branches with a tMRCA 25-30 generations ago. Otherwise connections between branches with surnames are essentially random. [28]
Illustration Nine provides a dendrogram of the entire group of FTDNA test kits for the L-497 Haplogroup work group. It includes testers who have minimally completed a Y37 STR test. The L-497 subclade, of which the Griff(is)(es)(ith) paternal line is a part, genetically branched off around 8900 BCE, the man who is the most recent common ancestor of this line is estimated to have been born around 5300 BCE. There are about 1,760 FTDNA based DNA tested descendants, and they specified that their earliest known origins are from Germany, England, United States, and 53 other countries. I included the entire group of test results to show the general shape and patterns revealed in the dendrogram.
STR distance dendrograpms usually contain clear and distinct clades, which are sets of men with a common ancestor. Such clades are characterized by a curved top boundary. in the dendrogram. This is what gives the dendrogram its characteristic ‘slope shape’. If we had test results of all family members the dendrogram would be more square shaped and resemble a family tree. Since that is impossible, there are obviously gaps and the sloping tops for respective clades of the dendrogram is due to the statistical range of the STR mutations and the history of a given haplogroup. .
While the G haplogroup was one of the dominant lineages of Neolithic farmers and herders who as a second wave into Europe, migrated from Anatolia to Europe between 9,000 and 6,000 years ago, they were overtaken by the R Haplogroup as part of a third wave of human migration into Europe and are consequently are presently a minority genetic group in Europe. The male lineages represented by the G haplogroup line are diminished and this is represented in dendrograms with long thin lines through time representing fewer male descendants.
I have highlighted distinctive clades in Illustration Nine as well as indicating the relative position of two possible descendants of William Griffis. To get a better view of this long Dendrogram, I have included a PDF version which allows one to increase the magnification of the image.
Illustration Nine: Dendrogram of FTDNA Y37 to Big Y Test Results for Members of the L-497 D-DNA Group

Click for larger View
If we look a bit closer at the results that are roughly highlighted in Illustration Nine, we can still see the “slope of an approximately family genetic clade structure” for individuals that have a Williams surname. This is reflected in illustration 10. My line of patrilineal descendants have a MRCA with this Williams clade around 14 generations ago. This MRCA was born would be about 434 years before present or about 1488 CE.
Illustration Ten: Dendrogram of FTDNA Y37 – Big Y Test Results for Members of the L-497 D-DNA Group – Blow-Up Portion Where My Test Kit is Located
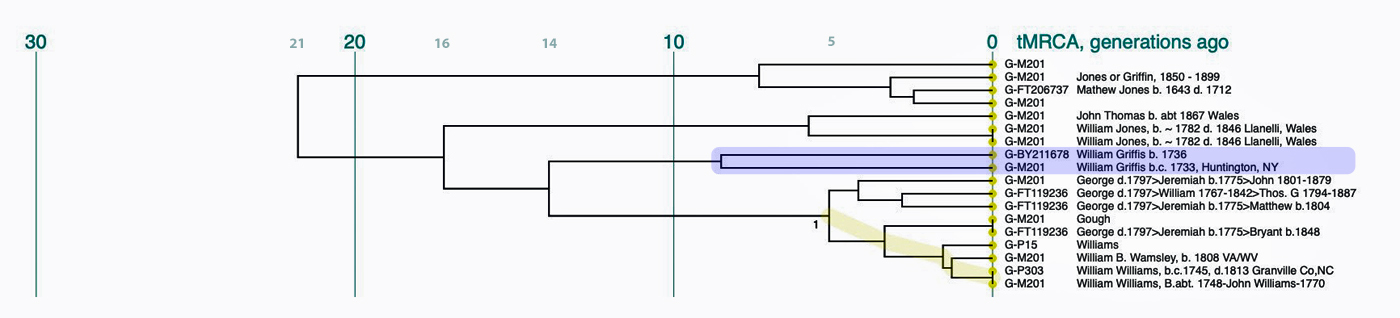
The dendrogram reinforced the connection with Mr. Griffith’s test kit. The dendrogram shows that we have a common ancestor about 8 generations ago. I highlighted our two kits in the dendrogram.
An alternative view of the dendrogram in Illustration Ten is provided by tightening the generational time scale, is provided in Illustration Eleven. It is the same data but the horizontal scale of the dendrogram has been shortened.
Illustration Eleven: Dendrogram of FTDNA Y37 – Big Y Test Results for Members of the L-497 D-DNA Group – Blow-Up Portion Where My Test Kit is Located, Shortened Time Horizontal the scale
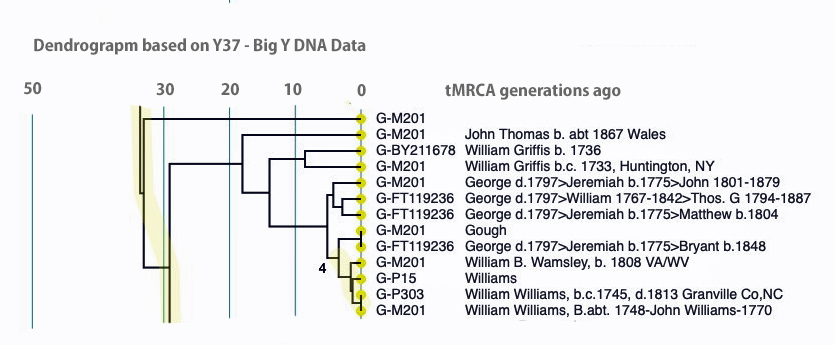
Click for larger View
Comparing the SAPP and dendrogram results with the Genetic Distance results reveal similarities. They both point to a genetic relationship with Kit 285458 (Griffith) with my Kit (285614). Both analyses point to a MRCA between our kits at 8 generations.
What’s Next
The next part of the story provides the results of corroborating a Griff(is)(es)(ith) relative, Henry Vieth Griffith, through the analysis of Y-DNA STRs!
Sources
Feature Image of the story is a dendrogram of comparing test kits results of Y-STR tests. Dendrograms are software-generated diagrams that convey relationships based on distance measured in generations. The dendrogram graphically portrays th genetic distance between individuals who are genetically related to me in the past 20 gnerations (e.g. the past 660 years). It is a graphic and mathematical confrmation of my conneection with Henry Vieth Griffith.
[1] Chang J (1999) Recent common ancestors of all present-day individuals. Advances in Applied Probability 31: 1002–1026.
Rohde DLT, Olson S, Chang JT (2004) Modelling the recent common ancestry of all living humans. Nature 431: 562–566.
Rohde DL, Olson S, Chang JT; Olson; Chang (September 2004). “Modelling the recent common ancestry of all living humans” (PDF). Nature. 431 (7008): 562–66. Bibcode:2004Natur.431..562R. CiteSeerX 10.1.1.78.8467. doi:10.1038/nature02842. PMID 15457259. S2CID 3563900
[2] Kevin P Donnelly, The probability that related individuals share some section of genome identical by descent. Theoretical Population Biology Vol 23: Issue 1, 1983, Pages 34–63. https://www.sciencedirect.com/science/article/pii/0040580983900047
[3] Rohde DLT, Olson S, Chang JT (2004) Modelling the recent common ancestry of all living humans. Nature 431: 562–566.
[4] John Hawks, When did humankind’s last common ancestor live? A surprisingly short time ago, 10 Jul 2022, John Hawks Weblog, https://johnhawks.net/weblog/when-did-humankinds-last-common-ancestor-live/
[5] Identical ancestors point , Wikipedia, This page was last edited on 17 December 2022, https://en.wikipedia.org/wiki/Identical_ancestors_point
[6] Genetic Distance, Wikipedia, This page was last updated 7 Dec 2022, https://en.wikipedia.org/wiki/Genetic_distance
Genetic distance, International Society of Genetic Genealology, Page was last updated 31 Jan 2017, https://isogg.org/wiki/Genetic_distance
Understanding Y-DNA Genetic Distance, FTDNA Help Center, https://help.familytreedna.com/hc/en-us/articles/6019925167631-Understanding-Y-DNA-Genetic-Distance
[7] The Most Recent Common Ancestor, International Society of Genetic Genealology Wiki, This page was last editd on 31 Jan 2017, https://isogg.org/wiki/Most_recent_common_ancestor
David Vance, Chapter 16, Estimating Ages to Common Ancestors, David Vance, The Genealogist Guide to Genetic Testing, 2020
[8] Haplotype, Wikipedia, This page was last edited on 11 February 2023, https://en.wikipedia.org/wiki/Haplotype
[9] Modal Haplotype, Wikipedia, This page was last edited on 6 April 2020, https://en.wikipedia.org/wiki/Modal_haplotype
[10] Ancestral Haplotype, International Society of Genetic Genealology Wiki, This page was last edited on 31 January 2017, https://isogg.org/wiki/Ancestral_haplotype
[11] Most Recent Common Ancestor, Glossary of Terms, FTDNA Help Center , https://help.familytreedna.com/hc/en-us/articles/4418230173967-Glossary-Terms-#m-0-12
Most recent common ancestor, International Society of Genetic Genealogy Wiki, page was last edited on 31 January 2017, https://isogg.org/wiki/Most_recent_common_ancestor
Most recent common ancestor, Wikipedia, page was last edited on 20 October 2022, https://en.wikipedia.org/wiki/Most_recent_common_ancestor
What is YFull’s subclade age methodology, page accessed 9 Aug 2022, https://www.yfull.com/faq/how-does-yfull-determine-formed-age-tmrca-and-ci/
The results and methodology used for determining ages from Big-Y SNPs can also be found in Iain McDonald’s U106 analysis. Read the PDF version at http://www.jb.man.ac.uk/~mcdonald/genetics.html which are updated several times a year.
Iain McDonald, Improved Models of Coalescence Ages of Y-DNA Haplogroups. Genes. 2021; 12(6):862. https://doi.org/10.3390/genes12060862
Poznik, G., Xue, Y., Mendez, F. et al. Punctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosome sequences. Nat Genet 48, 593–599 (2016). https://doi.org/10.1038/ng.3559 for PDF version: https://pure.mpg.de/rest/items/item_2307728/component/file_2307727/content
Shigeki Nakagome, Gorka Alkorta-Aranburu, Roberto Amato, Bryan Howie, Benjamin M. Peter, Richard R. Hudson, Anna Di Rienzo, Estimating the Ages of Selection Signals from Different Epochs in Human History, Molecular Biology and Evolution, Volume 33, Issue 3, March 2016, Pages 657–669, https://doi.org/10.1093/molbev/msv256
Kun Wang, Mahashweta Basu, Justin Malin, Sridhar Hannenhalli, A transcription-centric model of SNP-Age interaction, PLOS Genetics doi: 10.1371/journal.pgen.1009427 , bioRxiv 2020.03.02.973388; doi: https://doi.org/10.1101/2020.03.02.973388
Zhou, J., Teo, YY. Estimating time to the most recent common ancestor (TMRCA): comparison and application of eight methods. Eur J Hum Genet 24, 1195–1201 (2016). https://doi.org/10.1038/ejhg.2015.258.
Most recent common ancestor, International Society of Genetic Genealogy Wiki, page was last edited on 31 January 2017, https://isogg.org/wiki/Most_recent_common_ancestor
Most recent common ancestor, Wikipedia, page was last edited on 20 October 2022, https://en.wikipedia.org/wiki/Most_recent_common_ancestor
For specific information on history of the haplotree and related nomenclature, see also: International Society of Genetic Genealogy, Y-DNA Haplogrouptree 2019 – 2020, Version: 15.73 Date: 11 July 2020, https://isogg.org/tree/
YFull has a documented system to estimate SNP ages. This is how to get their estimate:
Go to YFull’s SNP search page; 2) Enter a SNP name and click the Search button; 3) A green hyperlink, labeled with a haplotree branch name (e.g., “R-L47”), should be displayed. Click on it; 4) You should now see a branch of the haplotree. Typically, this branch will have two dates: (a) The “formed” date is an estimate of when this branch began to diverge from its surviving siblings. (Extinct siblings are unknowable and therefore ignored.) (b) The “TMRCA” date is an estimate of when this branch’s surviving children began to diverge from each other. (Again, extinct lineages are ignored.)
[12] The GD estimates and estimated number of Generations is based on FTDNATiP™ Reports, Most Recent Common Ancestor Time Predictor based on Y-STR Genetic Distance
Understanding Y-DNA Genetic Distance, FTDNA Help Center, https://help.familytreedna.com/hc/en-us/articles/6019925167631-Understanding-Y-DNA-Genetic-Distance
Concepts – Genetic Distance, DNAeXplained – Genetic Genealogy,, Blog, 29 June 2016, https://dna-explained.com/2016/06/29/concepts-genetic-distance/
[13] J David Vance, The Genealogist Guide to Genetic Testing, 2020 , Chapter 5, https://www.amazon.com/Genealogists-Guide-Testing-Genetic-Genealogy/dp/B085HQXF4Z/ref=tmm_pap_swatch_0?_encoding=UTF8&qid=&sr=
[14] Ibid.
[15] These illustrations of the relationship between genetic distance and generations are from: David Vance, The Genealogist Guide to Genetic Testing, 2020 , Chapter 5
The statistical analyses were based on:
J. Douglas McDonald, TMRCA Calculator, Oct 2014 version, Clan Donald, USA website, Https://clandonaldusa.org/index.php/tmrca-calculator
[16] “For the Y chromosome these rates assume a 31 year generation.”
J. Douglas McDonald, TMRCA Calculator, Oct 2014 version, Clan Donald, USA website, Https://clandonaldusa.org/index.php/tmrca-calculator
[17] “The original FTDNATiP™ Report was based on research by Bruce Walsh, Professor at the University of Arizona, and his 2001 paper in Genetics. Walsh used a theoretical approach to model STR mutation rates and estimate when two people’s’ paths diverged in the Y-DNA haplotree. He used an infinite allele model, which theoretically accounts for markers mutating more than once, which can obscure the true mutation rate.”
Introducing the New FTDNATiP™ Report for Y-STRs, FTDNA Blog, 16 Feb 2023, https://blog.familytreedna.com/ftdnatip-report/
[18] Big Y Age Estimates: Updates and the Battle of Falkirk, FTDNA Blog, 9 Sep 2022, https://blog.familytreedna.com/tmrca-age-estimates-update/
Phylogenetic age estimation, otherwise known as “divergence dating,” has a long and rich history that began in the 1960s. Two general classes of methods have emerged: a strict molecular clock, and a relaxed clock. Sep 19, 2022, FTDNA Blog, https://blog.familytreedna.com/tmrca-age-estimates-scientific-details/
The Group Time Tree: A New Big Y Tool for FamilyTreeDNA Group Projects, FamilyTreeDNA Blog, 15 Feb 2023, https://blog.familytreedna.com/group-time-tree/
[19] Introducing the New FTDNATiP™ Report for Y-STRs, FTDNA Blog, 16 Feb 2023, https://blog.familytreedna.com/ftdnatip-report/
[20] David Vance, The Life of Trees (Or: Still Another Phylogeny Program),SAPP Tree Generator V4.25, http://www.jdvsite.com
Dave Vance, Y-DNA Phylogeny Reconstruction using likelihood-weighted phenetic and cladistic data – the SAPP Program, 2019, academia.edu, https://www.academia.edu/38515225/Y-DNA_Phylogeny_Reconstruction_using_likelihood-weighted_phenetic_and_cladistic_data_-_the_SAPP_Program
Y-DNA tools, International Society of Genetic Genealology Wiki, This page was last edited on 30 June 2022, https://isogg.org/wiki/Y-DNA_tools
Sennet Family Tree Blog, The SAPP is up and running: a phylogenetic analysis of Sennett surname project members, 8 May 2021, https://sennettfamilytree.wordpress.com/2021/05/08/the-sapp-is-up-and-running-a-phylogenetic-analysis-of-sennett-surname-project-members/
[21] Introduction to Distance Dendrograms, Tracking Back: A Website for Genetic Genealology Tools, experimentation, and discussion, http://scaledinnovation.com/gg/gg.html?rr=ddintro
Michael Drout and Leah Smith, How to read a Dedrogram, Wheaton college, https://wheatoncollege.edu/wp-content/uploads/2012/08/How-to-Read-a-Dendrogram-Web-Ready.pdf
Tim Bock, What is a Dendrogram, DisplayR blog, no date, https://www.displayr.com/what-is-dendrogram/
Dendrograpm, Wikipedia, page was last edited on 7 September 2022 , https://en.wikipedia.org/wiki/Dendrogram
Prasad Pai Hierarchical clustering explained, Towards Data Science, 7 May 2021, https://towardsdatascience.com/hierarchical-clustering-explained-e59b13846da8
Tom Tullis, Bill Albert, Hierarchical Cluster Analysis, in Measuring the User Experience (Second Edition), 2013 https://www.sciencedirect.com/topics/computer-science/hierarchical-cluster-analysis
Rob Spencer, Simple Distance Tree, Tracking Back – a website for genetic genealogy tools, experimentation, and discussion, 2023-01-28, ,http://scaledinnovation.com/gg/treeDemo.html
Rob Spencer, Family Tree and Y-DNA Simulator, Tracking Back – a website for genetic genealogy tools, experimentation, and discussion, http://scaledinnovation.com/gg/familySimulator.html
[22] Rob Spencer, Y STR Clustering and Dendrogram Drawing, Click on Discussion Tab, Tracking Back Click – a website for genetic genealogy tools, experimentation, and discussion, http://scaledinnovation.com/gg/clustering.html
[23] Introduction to Distance Dendrograms, Tracking Back: A Website for Genetic Genealology Tools, experimentation, and discussion, http://scaledinnovation.com/gg/gg.html?rr=ddintro
[24] Rob Spencer, The Big Picture of Y STR Patterns, The 14th International Conference on Genetic Genealogy, Houston, TX March 22-24, 2019, http://scaledinnovation.com/gg/ext/RWS-Houston-2019-WideAngleView.pdf Page 28
[25].Rob Spencer, Introduction to Distance Dendrograms, Tracking Back: A Website for Genetic Genealology Tools, experimentation, and discussion, http://scaledinnovation.com/gg/gg.html?rr=ddintro
[26] Rob Spencer, The Big Picture of Y STR Patterns, The 14th International Conference on Genetic Genealogy, Houston, TX March 22-24, 2019, http://scaledinnovation.com/gg/ext/RWS-Houston-2019-WideAngleView.pdf
[27] Rob Spencer, The Big Picture of Y STR Patterns, The 14th International Conference on Genetic Genealogy, Houston, TX March 22-24, 2019, http://scaledinnovation.com/gg/ext/RWS-Houston-2019-WideAngleView.pdf Page 12
[28] Rob Spencer, The Big Picture of Y STR Patterns, The 14th International Conference on Genetic Genealogy, Houston, TX March 22-24, 2019, http://scaledinnovation.com/gg/ext/RWS-Houston-2019-WideAngleView.pdf Page 11







{kind=link}
{kind=link}
{kind=link}