

I did not receive ground breaking results from my initial completion of autosomal DNA (atDNA) tests back thirteen years ago. Perhaps I did not totally understand and appreciate how to use the results provided by autosomal tests to the fullest. I think I was more interested in the ethnicity estimates produced by the atDNA than the actual matches with other possible living, distant relatives. When I completed the AncestryDNA test around 2012, autosomal DNA testing was at its commercial infancy. The database of completed tests was comparatively small so the DNA matches were not as notable as they are today. [1]
I had my father and his sister take the tests for my curiosity sake. I wanted to see how the test results differed between each of our tests. My genealogical research at that time was focused on the more traditional aspects of finding historical facts and evidence on various family lines of descent and information on specific individuals.
All this changed when I received a message on October 23rd, 2019. The test results led to a discovery of two half brothers! As an only child, I would at times wonder what it would be like to have brothers.
While I was dumbfounded and flummoxed by the discovery, I have now been gifted with having two brothers through this discovery. Further collaborative work with my half brothers have revealed how this connection unfolded in our lives.
While this news was earth shattering to me, my half brothers and for all families involved, each family has accepted and embraced the genetic revelations. The adoptive parents of Greg, who are still alive, are fully supportive of the three of us establishing family relationships.
Since my half brothers are alive, for purposes of privacy I have only referred to them by their first name in this story.
Discovering Siblings Through Genetic Testing
There are numerous anecdotal stories of people discovering biological family members through atDNA testing. These stories underline that interpreting DNA results often goes beyond sterile numbers. It involves navigating complex emotional territories where understanding ‘shared centimorgans’ can facilitate reconnecting lost, unknown, or separated family members. As such, a shared centimorgan is a powerful tool that can help piece together scattered familial puzzles, reveal hidden secrets and offer not just answers but also emotional closure for many. [2]
“Discovering “new” family members through DNA genealogy testing can trigger a wide range of emotions, including happiness, anxiety, sadness, or even anger. In fact, the emotional experience may be so intense that many genealogy sites state they are not liable for any “emotional distress” that may result from using the service.” [3]
According to studies on direct-to-consumer atDNA testing, a small but significant percentage of people discover they have a full or half sibling they were previously unaware of through their results. This makes it a relatively common occurrence, though not the majority experience for most users. While “sibling” is often used in this context, the discovered sibling could also be a half-sibling (sharing only one parent) [4]
In one study that attempted to gain an understanding of the prevalence of discoveries and associated experiences of atDNA testers, it was found that “most (82%) … learned the identity of at least one genetic relative. Separately, most respondents (61%) reported learning something new about themselves or their relatives, including potentially disruptive information such as that a person they believed to be their biological parent is in fact not or that they have a sibling they had not known about.” [5]
Adoptees often pursue genetic genealogy testing to find biological relatives. Another primary reason for atDNA testing is to gain insights from medical genomic testing. Adoptees understandably seek genetic medical testing for various reasons, primarily related to understanding their health risks and making informed decisions about their medical care in absence of knowing the medical histories of biological kin. They may choose direct-to-consumer testing because of its affordability and accessibility. [6]
Reaching Out and Revealing the Discovery
DNA testing companies typically offer internal communication platforms or features that allow users to reach out to potential DNA matches. While these companies provide communication platforms, users typically have control over their privacy settings and can choose whether to make themselves visible or contactable by matches. Additionally, the specific features and functionality of these communication platforms vary and can be subject to change as companies update their services. [7]
I received an AncestryDNA internal message on October 23, 2019 from David that contained information that not only were we half-brothers, but I had another half brother that was his full brother. It was a lot to mentally and emotionally digest!
October 23, 2019 ancestry.com Internal Mail Message

I thought David did a great job in succinctly conveying a number of points surrounding his discovery. He got straight to the point with the news. He was empathetic to my situation of receiving this news. He also made sure reaching out to me would not cause any ripples in my family. Since his adoptive parents and my parents had passed, he considered the timing of reaching out to me.
David indicated that all the revelations of his having a full and half brother came to light within a three month time period. His discovery of our relationship was the result of completing an AncestryDNA test after he completed a 23andMe test where his full brother Greg discovered the relationship with Dave.
My Immediate Reaction
I was waiting for the car to warm up on a cold fall morning and was quickly going through messages I had received in the night. I was preparing to drive to a remote area for a morning gravel cycling ride. I rarely receive notices from AncestryDNA so Dave’s message caught my eye. I read and reread David’s message a few times. I sat in the car rereading the message for about ten minutes. I decided to digest what I had read on my bike then reach out to Dave when I got home.
I had many thoughts swirling in my head, trying to reconcile potential facts with family history and my father’s colorful life. I was trying to fit all of this together. For my father to have two children from the same person and then give them up for adoption was racking my brain and heart.
At the same time I could only imagine what he must have been going through to follow through this process. We do not know and will never know. He took this part of his life to the grave. I only can make conjectures on what happened and why, given what his life was like at the end of the 1950’s and early ’60’s. I could imagine that he was clearly boxed in by his actions and the subsequent demands placed on his life. Perhaps in his view, his only recourse was to help with the births and adoption. Otherwise the life he knew would have been torn asunder.
My father and mother married when they were 20 and I came along within that year. He was trying to finish college, adjusting to married life, and caring for a family. He was living in a new world full of responsibilities, economic challenges and social pressures. I know that during his 20’s and 30’s, my father enjoyed living in two worlds, one associated with being a father and husband and the other world which was on the edge, staying out late gambling, playing cards and betting on horses and associating with a ‘different crowd’. I witnessed many arguments as a child, not really knowing what the adults were fighting over.
Over time my father became my best friend and best man in my weddings. Since the time I had a ‘consistent paying’ job in the early 80’s, I had called my father at lunch or after work everyday. The calls could have been 30 minutes or a short minute just to say hello. They became part of our ritual. I considered it unique and special to have a best friend and father all wrapped up into one.
While best friends always have secrets, I figured I knew my dad’s past fairly well. I was aware of the good and the not so good in his life.. He had a successful career in sales, was an accomplished regional master’s tennis player, started his own business, and immeasurably helped his family and friends in many ways throughout his life. He had a huge heart and like many, made a few mistakes along the way. His trajectory through life was full of twists and turns. My father had many facets to his life. This was my father that my family knew.
Having two children out of wedlock was certainly a surprise. However, having two children with the same person was more perplexing for me. This reflected something more than a fling or brief encounter. I also wondered but could understand why he never discussed this part of his life to me or others. While I was trying to make sense of this, I looked forward in getting more information from my newly found brothers, Dave and Greg, to figure it out.
When I returned from my bike ride, I wrote an email to Dave full of questions. He was genuinely happy to hear from me. We both harbored no ill will or bad feelings. We both wanted to simply obtain a clear, objective picture of the narrative, he from the adoptive side; and me from the revelation that I have brothers from another relationship of my father’s.
The DNA Results
When I conveyed to my extended family that I had discovered two half siblings through DNA testing, one of my relatives asked, “How do you know if the DNA tests are accurate or legit?“. My direct answer was the results were accurate. My answer, however, was based on both genetic knowledge and also traditional genealogical sleuthing for facts.
Between the three of us, we completed atDNA tests with 23andMe and AncestryDNA. All three of us completed tests with 23andMe. The only DNA test we do not have is an AncestryDNA test for Greg. This test would document the genetic relationship between our father and Greg.
As stated previously, I as well as my father and paternal aunt completed autosomal tests seven years prior to Dave’s discovery. Having my father at the time complete an atDNA test provided prescient knowledge about our family genetics. Dave’s initial AncestryDNA test results indicated that ‘jimgriffis’ was his biological father and that my paternal aunt and I were close family members, possibly first cousins. (See illustration one.)
Illustration One: Dave’s AncestryDNA Autosomal Results

“Half-siblings on Ancestry DNA will show up as “Close Family” or “First Cousins” and are expected to share an average of 1,759 centimorgans with a range of 1,160-2,436 centimorgans, according to data from the Shared Centimorgan Project.” [8]
Half-siblings typically share approximately 25 percent of their DNA, between 1,160-2,436 cMs, and unlike full siblings, do not share fully identical regions (FIR). [9] There is a bit of an overlap of shared cMs for a number of genetic relationships in this cM range. If you do not have other forms of genealogical information, half-sibling DNA patterns can be confused with niece/nephew relationships, aunt/uncle relationships, and grandparent/grandchild pairs.
“When interpreting autosomal DNA statistics, one must be careful to distinguish between the distribution of shared DNA for given relationships and the distribution of relationships for given amounts of shared DNA.” [10]
This overlap is reflected in a genetic relationship chart produced by the Shared cM Project [11], see illustration two below. I have used a cM value of 1722 and 1735 since the atDNA shared cM test value with me for Dave is about 1735 cMs and with Greg is about 1722 cMs for the 23andMe test results. [12]
Illustration Two: Possible Relationships with a cM Value of 1722 and 1735
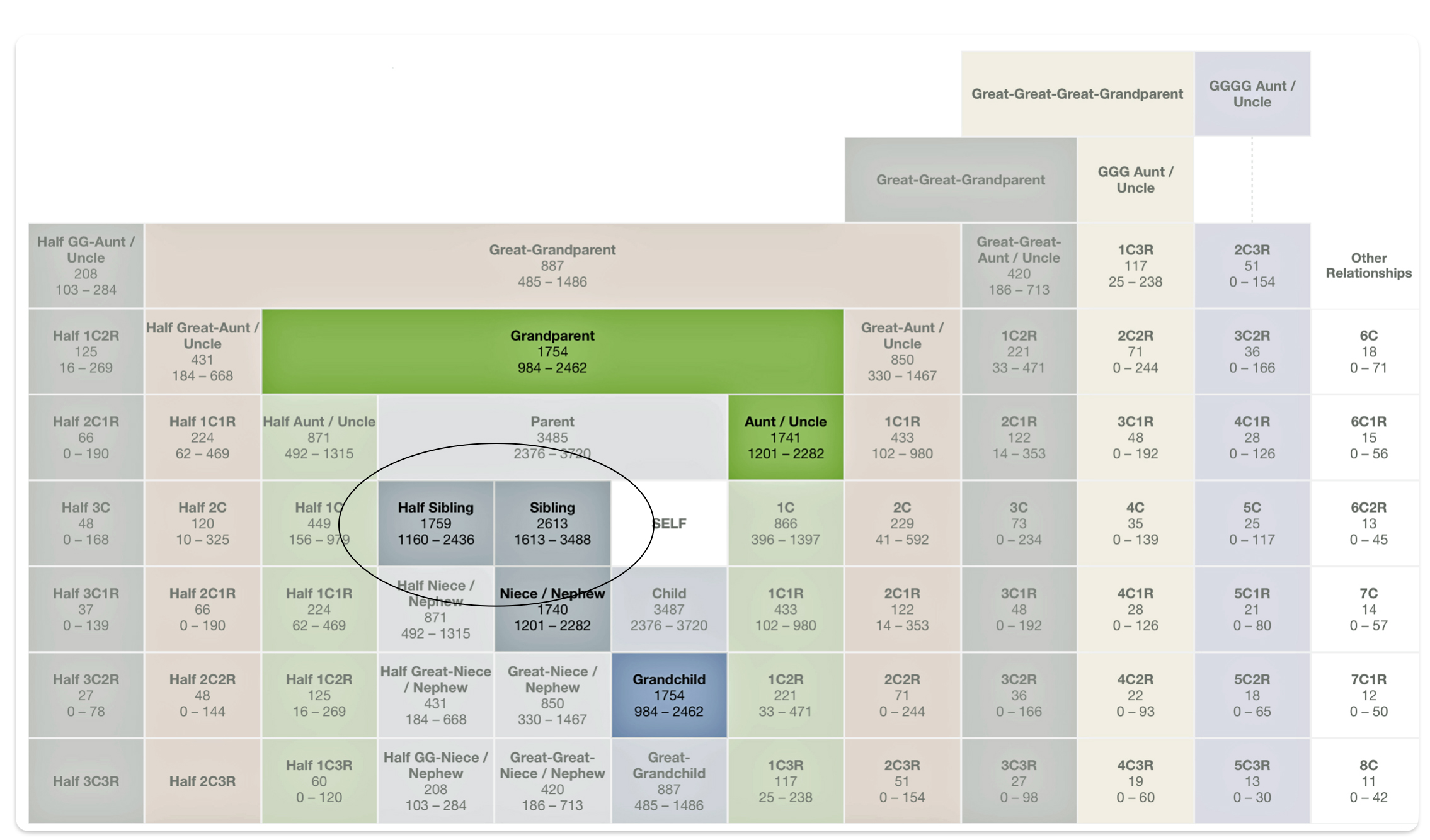
Aside from the possible relationships that can be found with a cM value of 1722, it is interesting to note the overlap between sibling and half-sibling relationships in illustration two. The cM range for siblings is 1313 – 3488 and the range for half-siblings is 1160 – 2436, with an over lap of 823 cMs.
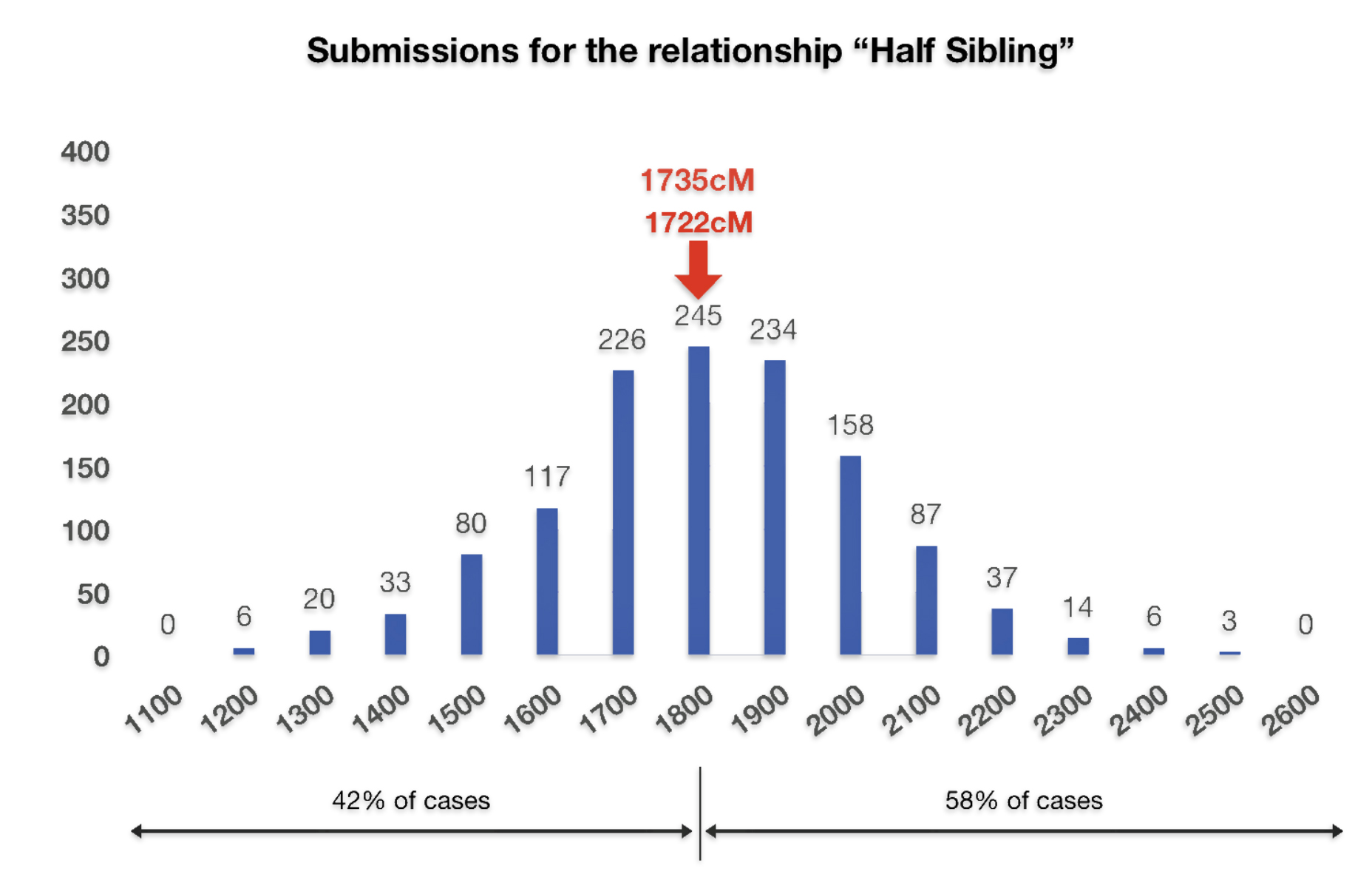
If we look at the total number of submissions in the Shared cM Project for ‘half-sibling’ relationships, there were 1266 submissions for the Half Sibling relationship with a mean value of 1759 cM and a standard deviation of 207cM. Illustration three provides the distribution frequency of the cM values for half-sibling matches. Basically, a value of 1722 or 1735 is hovering around the middle of the distribution of cM values for half siblings. Hence, my answer to the question of the legitimacy of the test results is the results are pretty solid and reliable.
Illustration Three: Distribution of cM Values for Half Sibling Relationships in the Shared cM Project
The cM test results for matches can differ between DNA companies. For example table one below reflects the estimated cM values for matches between me and my half brothers based on AncestryDNA and 23andMe test results. Both companies report results in different ways. Depending on the DNA company, the predicted relationship is depicted by different measures: the total percentage of shared DNA, the number of shared segments, the length of the shared segments, the longest block of cMs. Different companies may also provide slightly different relationship estimates due to variations in their testing algorithms and reference databases.
Essentially 23andMe provide percent of shared cMs and AncestryDNA provides number of shared cMs to document genetic relationships.
Table One: cM Match Results between Jim, Dave and Greg
| cM Share Half-Sibling Relationship with Jim | Percent Shared cM (23andMe) | AncestryDNA Number of matched cMs | cM Conversion using Shared cM Project conversion | Conversion using 68 x % Shared |
|---|---|---|---|---|
| David | 23.32 | 1685 | 1735 | 1585.76 |
| Greg | 23.14 | – – | 1722 | 1573.52 |
The cM ranges for each of the DNA companies and the Shared cM Project also differ, as reflected in table two.
Table Two: cM Ranges for Half Sibling Relationships
| Source | cM Range for Half Sibling |
|---|---|
| 23andMe | 1264 – 2529 cM |
| AncestryDNA | 1450 – 2050 cM |
| Shared cM Project | 1160 – 2436 cM |
When Dave notified me of our genetic relationship, I revisited and reviewed my DNA matches in AncestryDNA. I had not reviewed my matches in a long time; and there was Dave as a half brother!
The number of shared cMs between my father were similar to the results Dave received in his test results. I shared 3,479 cMs across 26 segments with my father ‘jimgriffis’. Dave shared 3,464 cMs with ‘jimgriffis’ across 57 segments. (See illustrations Three and Four.)
Illustration Four: My AncestryDNA Autosomal Matches
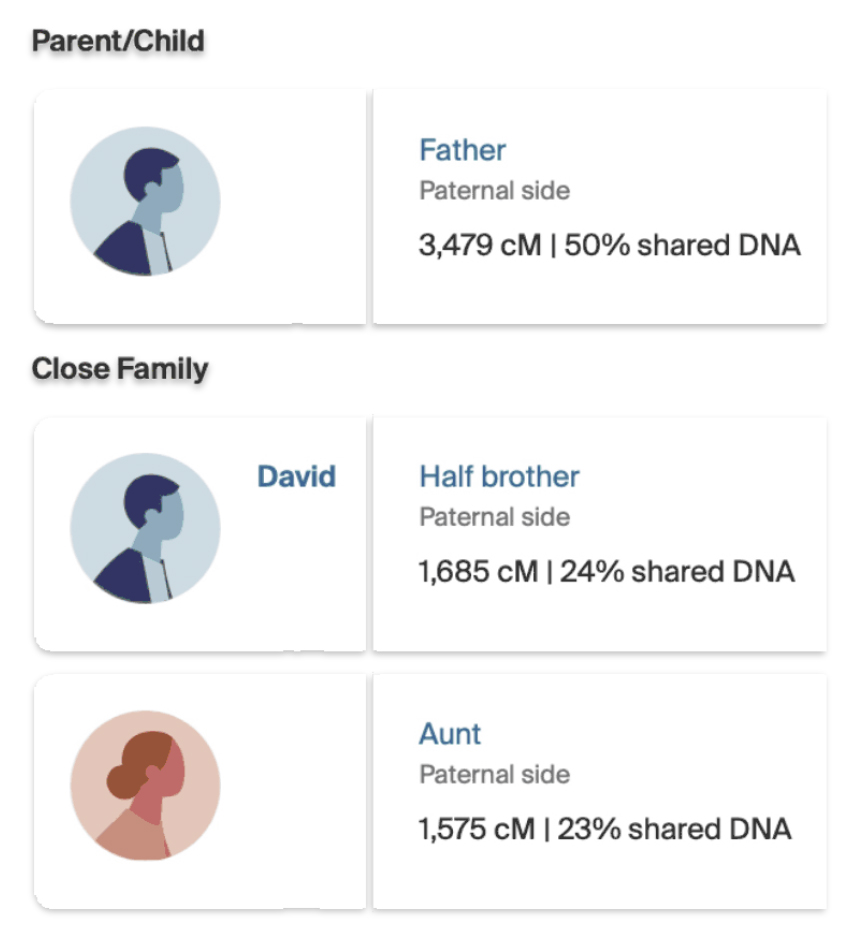
Both Dave and I also have similar matches with our paternal aunt. I share 1,575 cMs and 41 segments with my paternal aunt. Dave shares 1655 and 52 segments with our paternal aunt. The ancestryDNA numbers are within the cM range for an aunt/nephew relationship, as reflected in illustration five..
Illustration Five: Shared cM Project Submissions for Aunt/Uncle
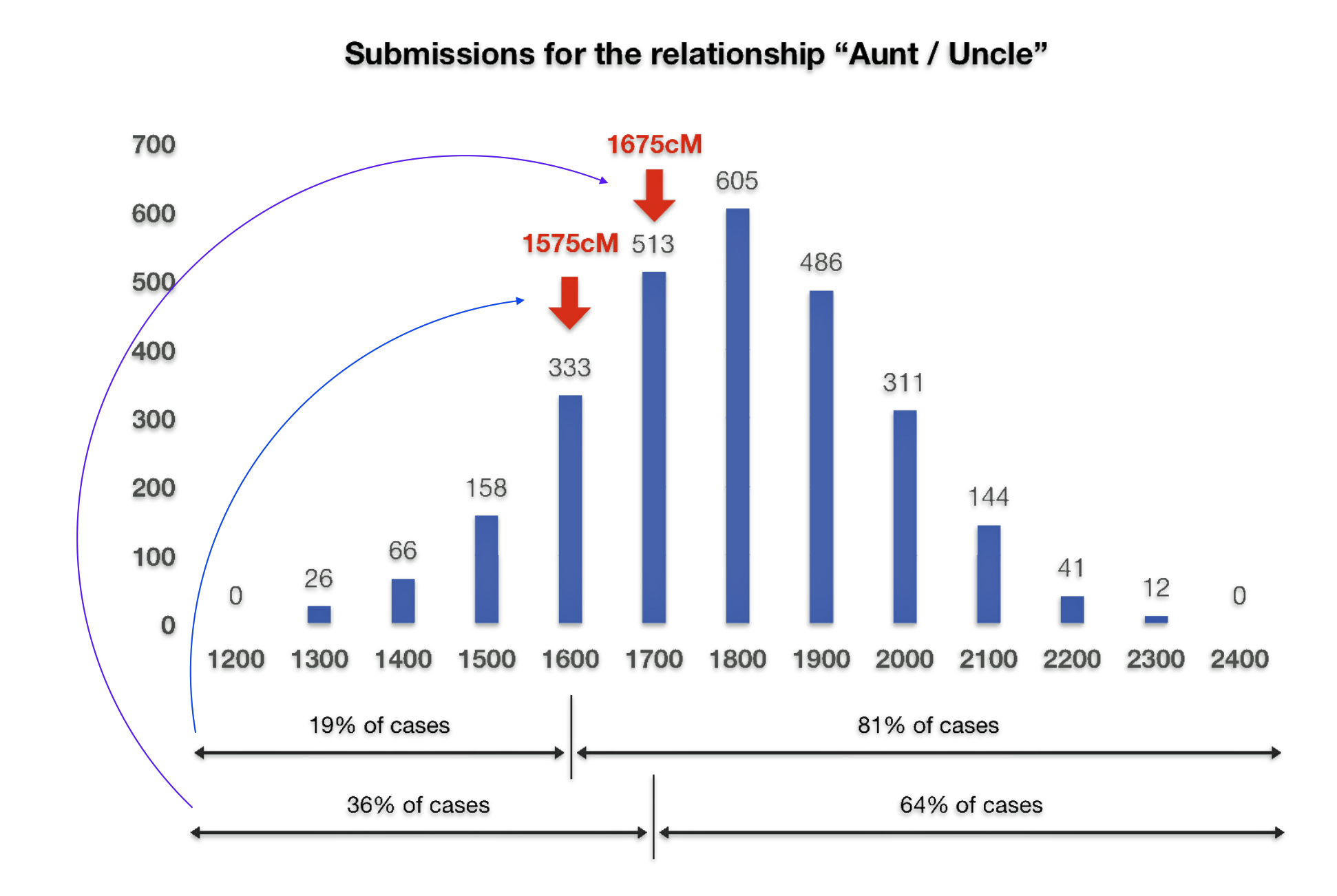
At the beginning of November 2019, I completed an atDNA 23andMe test to validate the DNA connection between the three of us. Before completing the test, I only had a test connection with Dave. The following are the results of my 23and Me atDNA test.
Illustration Six: 23andMe Autosomal Matches

As reflected in illustration six above, the numbers are very close for each half sibling relationship.
Based on the science, half-sibling DNA relationships show distinct patterns that can be reliably identified through atDNA testing. Modern DNA tests can achieve up to 99.9% accuracy for half-sibling relationships when confirming shared centimorgan (cM) ranges, using tests that analyze hundreds of thousands of DNA markers, and including the known parent’s DNA in testing. [13]
As the youtube video below discusses, atDNA tests can identify half-siblings with a high degree of accuracy, additional relationship testing or analysis may sometimes be needed for full confirmation, especially in complex cases. The tests are generally very reliable for distinguishing half-siblings from full siblings or unrelated individuals.
Can atDNA Tests Find Siblings or Half Siblings?
“(A) DNA test can prove half-siblings. As a matter of fact, it’s the only accurate way to establish the biological relationship between the people in question. In a half-sibling situation, the siblings share one biological parent. But you need to test the parent. Here is are the steps involved:
- The potential half-siblings need to share 1160-2436 cm.
- If the potential siblings are in range AND share more than 1600 cm, there must not be any fully identical regions. If there are, then it’s more likely a full sibling relationship.
- Each potential half-sibling must share 2500-3720 cm with the parent.
If all three steps are true, then you’ve got yourself a half-sibling relationship.” [14]
Background of the Full Brothers’ Discovery and Research
Similar to many individuals who were adopted, Dave and Greg completed atDNA tests to understand their medical predispositions and fill gaps in their family health history, which becomes increasingly important as one ages and have children of their own. This information helps providing answers to routine medical questions about hereditary conditions and genetic risks that doctors typically ask during examinations.
In addition to exploring genetic health history, Greg had been trying to find out more about his biological past since 2007. He knew from his adoptive parents that he was born in Rochester, New York. The adoption agency in Rochester sent him a note back in 2007 that stated his father was a salesman, married, and his mother was a nurse. Both were college educated. The father helped with the costs of birth and adoption. The note stated that ‘both parents were very religious and the controversy would have been too much so adoption was the solution‘.
In the summer of 2019 Greg discovered Dave as a full brother from a 23andMe match after Dave completed the test. Similar to Greg, Dave also completed the 23andMe test at his wife’s behest, to gain knowledge about his genetic medical past. At the time, Dave said he did not have much faith in the results.
Greg reached out to Dave on July 29, 2019 with “Hello Bro” as the subject line in an email. This started the ball rolling. According to the 23andMe analysis, they both are full siblings.
After this email, Dave started a concerted effort at obtaining additional information about his biological past. Greg had been conducting research previously and tracked their mother, Esther, to Arizona and her marriage in 1973 and her subdeath in 1996. Esther was a nurse by profession. Her nursing career took her many places, from Albany, New York to New Haven, Connecticut, to Alaska, and then to Phoenix, Arizona where she was married and had three sons. Through their collective efforts, Dave and Greg discovered four half siblings!
Using various sources, Dave started to piece together Esther’s family who was originally from the Kingston, New York area. He found a friend of Esther’s and nursing school classmate of Esther’s named Phylis Hutton. Both started their nursing careers in Albany, New York.
When Dave discovered Phylis in 2019, she was in her 80’s and living in Kingston, New York. He had a short telephone conversation with her. She indicated she remembered and knew about Esther going to Pittsfield to have a child. Dave was born in Pittsfield, Massachusetts. Dave asked if she knew of the father. She said she did not remember the name but she recalled that his father was a reverend and recalled that he was an orderly at the hospital and that ‘he was extremely handsome’.
Newspaper Announcement – Esther Emerick and Phyllis Hutton

A short time after his call with Phyllis, Dave received another call from a newly found first cousin from his biological mother’s side. His cousin was contacted by Phyllis about the news and her telephone call with Dave. Dave’s first cousin then received information from another cousin and advised Dave to follow up on a name ‘James D. Griffis’ from Troy, NY. The cousin stated that his father was Harold W Griffis, a prominent minister back in the 1950s-1960s. It was thought that James was Esther’s suitor at the time and that James had a brother John and a sister.
In early August 2019, Dave received his pre-adoption birth certificate. The father was not listed but his mother was listed as Esther Emerick, born 1938, Kingston, New York.
Working Together: Verifying Facts, Time and Place
At the time Dave initially reached out to me, the historical information regarding their biological father did not entirely jibe between Greg and Dave’s research. The biological father on both Dave and Gregs’ adoption forms indicated that their father was a salesman. Phyllis Hutton, from Esther’s nursing past, indicated that the father was an orderly at the hospital that she and Esther were employed in Albany. Before his passing, Dave’s adoptive father indicated that he knew his biological father was a salesman and his biological mother was a nurse.
To obtain additional or potential new leads, Dave completed an ancestry.com DNA test to see if people would show up as close relatives. He received his results October 17, 2019. He opened the results and looked at DNA matches on October 21, 2019. I, Nancy, and my father showed up as close relatives. Dave then sent me the note on the 22nd of October.
After a few email exchanges, Dave and I scheduled a telephone conversation about a week after his initial contact with me. We had a two hour conversation on many subjects. One part of the conversation, tied the facts and events together.
My dad was a salesman but he also had a second job as an hospital orderly around 1959 – 1961. This would explain the discrepancy between the stated occupation on the adoption documents and oral history that was obtained from Esther’s family and friend.
I recalled my father working nights when I was in first grade. I recall one time meeting my father with my mother in our car one morning near a big brick building which was the Albany Medical center. The adults were talking. but I paid little attention to what was discussed. As a child in the back seat of the car, I recall my Dad leaning into the window as he was standing beside the car. I did not listen but I recall my Dad saying at the end of teh conversation, “Well, I need to go to my other job now” and they said their goodbyes and my mother drove on to do errands.
When my father got married, my paternal grandparent’s ‘social contract’ with my father was that they would financially help him with college until he got married. Once he got married, he was on his own, he had to pay for his own education and living costs.
My parents married when my father was a Junior in college. My dad subsequently worked two jobs to support a family and school costs. I was born while he was in college. One of his two jobs was working as an hospital orderly in the state mental institution on the night shift while he finished college.
After graduation, he and his young family moved back to the Troy, New York area. He continued his colorful ways. He accrued a lot of debt probably through gambling. He received financial assistance from his brother’s father-in-law who was a banker.
I believe this was a melting point for him, for my mother, and his parents Harold and Evelyn. He needed funds to supplement his current standard of living associated with his day job to pay off the debt. He again got a night job based on the skill sets he knew he had – being an orderly at Albany Medical.
I told all of this to Dave and asked when and where his mother was a nurse in Albany. Dave directed me to the newspaper article above. She was nurse at Albany Medical in 1959-1960. Phyllis’ story started to make sense. My dad was a salesman at Kimmey Company, a plumbing construction company, and also an orderly at Albany Medical.
Dave was born in 1960 in Pittsfield, MA. His brother Greg was born in 1961 in Rochester, NY. Based on information gleaned from Dave and Gregs’ adoption papers, our father was fully aware of their births and it appears he provided financial support to Esther in the birth and adoption process.
Non-Marital Pregnancies and Adoptions in the late 1950s and early 1960s
Having and keeping a child out of wedlock would have been quite a challenge for Esther. Esther was from a ‘prominent religious oriented’ family in Kingston, New York. The possibility of raising two children out of wedlock was inconceivable. James had a young family, was married to a practicing Catholic wife and was raised by a Methodist minister. Based on their upbringing, abortion was not a moral choice for both Esther and James even if it were legally available at the time. For various unknown reasons on both sides, divorce was not an option as well.
Esther was starting her nursing career in 1959. To have two children and be a single mother back to back in 1960 and 1961 would have been daunting. I can only imagine the stress and social and economic challenges that must have been placed on our father, Esther and my mother. I do not know if my mother was aware of the births. I assume that she was aware.
This was a period in American history that was known as the Baby Scoop Era. It started after the end of World War II and ended in the early 1970s, characterized by an increasing rate of pre-marital pregnancies over the preceding period, along with a higher rate of newborn adoption. [15]
“It was common knowledge that many white unwed mothers had the resources to conceal their pregnancies, often by traveling far from home to have their babies, to states that didn’t record illegitimacy on birth certificates. ” [16]
The ability to avert having a child in the late 50’s was difficult given the limited options for contraception and the legal and religious prohibitions placed on abortion. For non-wed mothers, the viable option was having the child and offering the child up for adoption.
The legal status and accessibility of birth control was severely restricted by the 1873 Comstock Law, which criminalized contraceptives and banned their distribution through mail or interstate commerce. [17]
In the 1950s, “Americans spend an estimated $200 million a year on contraceptives. Due to massive improvements over the past decade in condom quality and a growing awareness of the inadequacies of douches, “rubbers” are the most popular form of birth control on the market.
“Although the vast majority of doctors approve of birth control for the good of families, anti-birth control laws on the books in thirty states still prohibit or restrict the sale and advertisement of contraceptive devices. It is a felony in Massachusetts to “exhibit, sell, prescribe, provide, or give out information” about them. In Connecticut, it is a crime for a couple to use contraception.” [18]
The year that Dave was born, “(t)he adoption of the birth control pill grew rapidly after its FDA approval on June 23, 1960. 400,000 women sought prescriptions in the first year, despite the high cost of $10 (equivalent to $80 today).” [19] The first pill, Envoid, in addition to prohibitive cost, it also produced some negative side effects: nausea in the first few months and weight gain.
In the 1950s, abortion was heavily restricted across the United States with severe consequences for both providers and women seeking the procedure. By 1950, abortion was illegal in every state except when necessary to save the woman’s life. Forty-four states only permitted abortion when the woman’s life was endangered. [20]
By the middle of the twentieth century, almost every state in the country had brought their adoption laws into alignment with the principles laid out by two influential groups: the U.S. Children’s Bureau (USCB) and the Child Welfare League of America (CWLA) guidelines and the Child Welfare League of America. [21]
“The USCB was created by the federal government in 1912 as a Progressive Era organization that introduced public health interventions to reduce infant mortality. It also became a national leader in making policy related to illegitimacy and unmarried mothers. The group was motivated by multiple scandals with commercial and unregulated adoptions that had lethal consequences for the infants. The CWLA, which brought together public and private service groups starting in 1915, later initiated efforts to standardize adoptions that culminated with its influential 1958 publication Standards for Adoption Service.” [22]
By the late 1940s, existing service organization like the National Florence Crittenton Mission—later called the Florence Crittenton Association— encouraged single mothers it served to relinquish their infants. It is not known if Dave and Greg were born in a Maternity home in Pittsfield and Rochester. [23]
Figures vary for the number of adoptions during the postwar decades, since most of them went unrecorded. One source indicates the number of adoptions more than doubled (128 percent) from an estimated 50,000 in 1945 to 114,000 in 1961, the year that Greg was born. As reflected in illustration seven, Dave (born 1960) and Greg (born 1961) were born and adopted in a period where there was a steady rising nationwide wave of adoptions. [24]
Illustration Seven: Adoption Trends 1944 – 1961
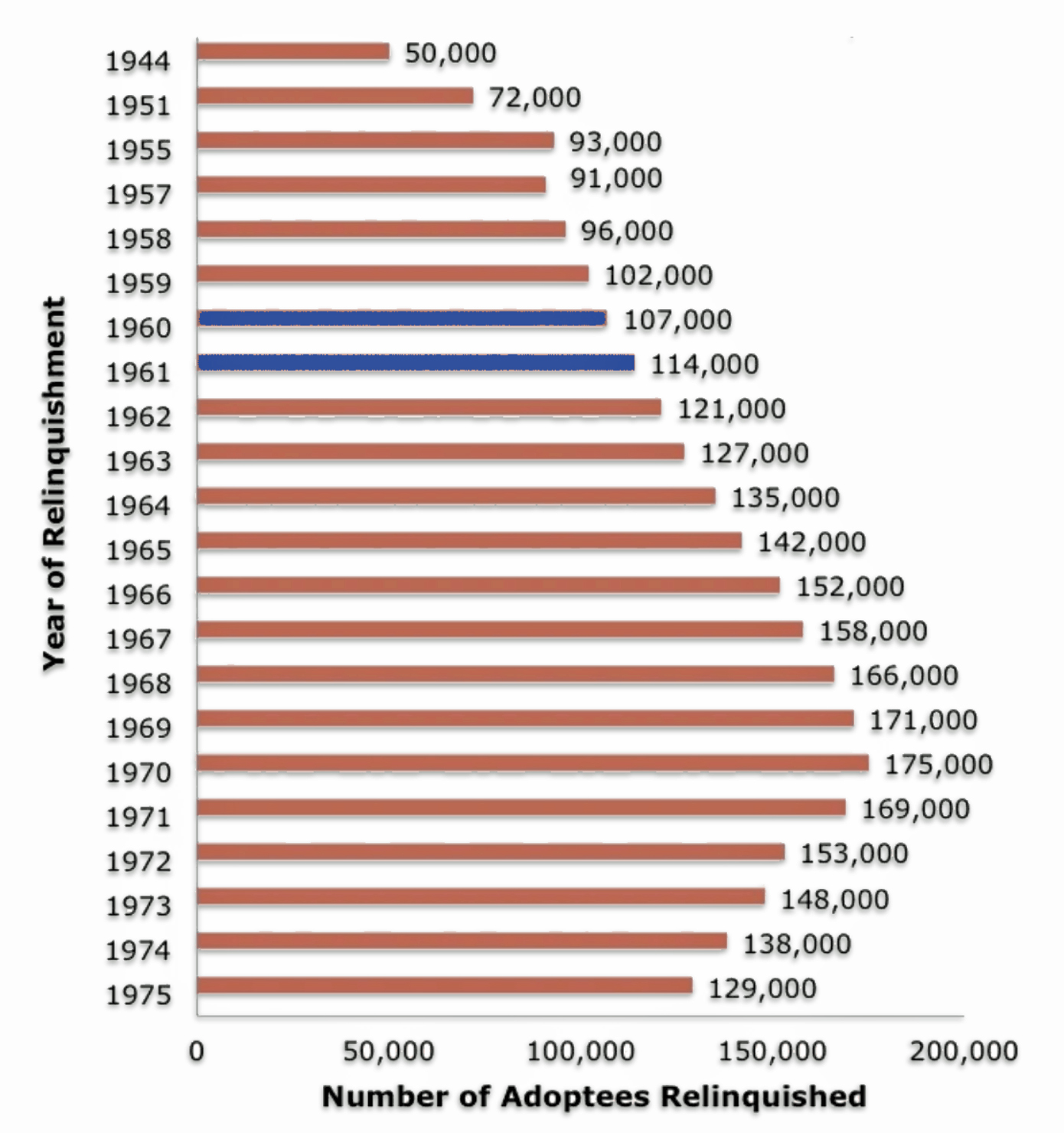
“The heyday for domestic adoption was the mid-20th century. Between 1940 and 1964, the rates of so-called “illegitimate” pregnancy doubled and tripled, from 89,500 in 1940 to 275,700 in 1964.” [25]
Family Support
Without judgement, I shared the surprising discovery of having two half-siblings to my immediate and extended family in an e-mail. I concluded with the following:
“Each of you had a unique relationship with my father. Based on that relationship, I am confident to state that he treated you like, as he would say, aces. He was there for you and loved you dearly even when there were hard times. So I hope before you cast judgment, as he would do, you see through your heart and then open your arms.“
I told Dave and Greg that I was very happy that they have found answers to questions that I imagine adopted children always have in the back of their minds. I told them I hoped they have or had wonderful parents and their life was good. I also told them I welcomed them as my brothers and looked forward to having them in my life. As an only child I always longed to have brothers.
Many of my family members replied to this news. One of my aunts indicated, “Jim was a young wild character and got into a lot of trouble with his marriage to Peggy, gambling and finances, and his relationships with other women. He grew up the hard way over his lifetime and became the loyal person we love and admire. He extended his care and loyalty to all of us.”
One of my cousins said in a reply to my email:
“I love that this family can accept this type of news- hold Uncle Jim accountable for the affairs, but forgive, love, and move forward, free of judgement. We truly are a class act clan!“
My cousin’s sentiment sums up the common sentiment I have received from family members. We as a family are strong, tolerant, have a good sense of humor and full of different personalities. We all are not perfect and if someone takes a misstep, we help them regain their balance. When you are connected by so much family and love, forgiveness and collectively moving on in life is much easier.
I know David and Greg appreciate the positive support. In addition to our communication, I have provided copies of a commemorative book I created that chronicles our father’s life. The book gives them an idea of what their biological father was like throughout his life.
Conversely, I have been introduced to many of Dave’s friends and his family on ‘his turf’. I have also have communicated with Greg’s step-parents who are proud and happy that we have found each other.

Moving Forward and Continuing the Journey
Discovering siblings late in life can be a complex and emotionally charged experience, involving a mix of excitement, confusion, curiosity, and sometimes even grief, as individuals grapple with a new family dynamic, a revised understanding of their identity, and the potential for a significant relationship that was previously unknown. This can be influenced by the circumstances surrounding the discovery, like adoption, family secrets, or a parent’s hidden past, leading to varying levels of adjustment and impact on personal relationships.
For Dave, Greg and me, I think we handled the discovery with excitement, gratitude and promise. We are in agreement that it would have been nice if we were able to experience having our brothers in our lives when we were in earlier stages of our lives. In absence of the shared past, we are grateful to presently have each other in our lives.
We discovered our relationship as siblings when we ranged in ages of 59 to 66. We do not have shared histories as children, adolescents, young adults, and when we went through mid-life experiences. We were not there for each other through our ups and downs. Our bond lacks all those experiences of ‘growing up’. Our bond is based on our unique past and the future, learning about each other’s past life and our respective families, and presently enjoying our time together as brothers.
A Zoom Call
Since the beginning of 2020, we have been attempting to arrange a time when all three of us can get together. Sad to say our schedules have not yet been able to coincide. As twosomes, we all have gotten together on various occasions. We do not live close to each other and we each have family demands. My two younger brothers are still working so they have the added demands of work life. I am confident the three of us will enjoy time together in the future and be part of each other’s lives.
Dave and Jim September 2021

Jim and Greg Thanksgiving Weekend 2024

Echoing a title of a book of an adoptee’s journey through the American adoption experience: ‘You don’t know how lucky you are!“. [26]
I think this statement is true for Dave and Greg … as well as for me.
Sources
Feature Image: This is a modified version of an illustration from Pereira, Rita, Pietro Biroli, Stephanie Von Hinke, Hans Van Kippersluis, Titus Galama, Niels Rietveld, and Kevin Thom. 2022. “Gene-environment Interplay in the Social Sciences.” OSF Preprints. 4 March 2022 DOI:10.31219/osf.io/d96z3; and a stock photo https://stock.adobe.com/
[1] Autosomal DNA testing has undergone significant changes and improvements since its introduction in 2009. 23andMe launched the first autosomal DNA test for genealogy in late 2009, marking a revolutionary change in genetic genealogy. This test allowed people to examine DNA inherited from all ancestral lines.
Family Tree DNA launched their Family Finder test in February 2010. AncestryDNA began rolling out their autosomal DNA test in the autumn of 2011, with an official launch in the United States on May 3, 2012. They initially kickstarted their database by offering free tests to over 10,000 selected subscribers. AncestryDNA reached 2 million users by August 2016.
The database showed exponential growth until April 2018. Growth slowed after April 2018, adding 6 million people instead of the projected 12 million in the following year. Database growth declined by 51% from April 2018 to May 2019. By 2021, AncestryDNA led the pack in database size with over 20 million completed test kits.
By 2014, AncestryDNA’s database had grown rapidly, selling 30,000 to 50,000 DNA kits monthly. The test became available internationally when AncestryDNA launched in the UK and Ireland in 2015, followed by expansion to 29 additional countries in February 2016.
Testing accuracy has improved significantly over time. Early ethnicity estimates were often inaccurate. Current continental-level results are now highly reliable.
Genealogical DNA test, Wikipedia, This page was last edited on 18 November 2024, https://en.wikipedia.org/wiki/Genealogical_DNA_test
History of genetic genealogy, International Society of Genetic Genealogy Wiki, This page was last edited on 27 April 2024, https://isogg.org/wiki/Timeline:History_of_genetic_genealogy
Doriottt, Candace, Genetic Codes Unraveled: New Clues to Human History. Ancestry magazine, January/February 2000, Page 15 – 21
Theunissen, C.A. The Effects of DNA Test Results on Biological and Family Identities. Genealogy 2022, 6, 17. https://doi.org/10.3390/genealogy6010017
AncestryDNA at Back To Our Past, 12 Nov 2014, Cruwys News, https://cruwys.blogspot.com/2014/11/ancestrydna-at-back-to-our-past.html
Williams, Ed, Analysis of AncestryDNA Tests Processed from June 2016 to August 2019, 12 Dec 2019, Counting Chromosomes, https://countingchromosomes.com/blog/70-analysis-of-ancestrydna-tests-processed-from-june-2016-to-august-2019
Venner, E., Patterson, K., Kalra, D. et al. The frequency of pathogenic variation in the All of Us cohort reveals ancestry-driven disparities. Commun Biol 7, 174 (2024). https://doi.org/10.1038/s42003-023-05708-y
Genealogical Database Growth Slows, 22 Jun 2019,The DNA Geek, https://thednageek.com/genealogical-database-growth-slows/
AncestryDNA Surpasses 20 Million, 27 May, 2021, The DNA Geek, https://thednageek.com/ancestrydna-surpasses-20-million/
[2] See for example:
Catherine A. Ball, Mathew J Barber, Jake Byrnes, Peter Carbonetto, Kenneth G. Chahine, Ross E. Curtis, Julie M. Granka, Eunjung Han, Eurie L. Hong, Amir R. Kermany, Natalie M. Myres, Keith Noto, Jianlong Qi, Kristin Rand, D. Barry Starr, Yong Wang and Lindsay Willmore, AncestryDNA Matching White Paper, Updated July 15, 2020, AncestryDNA, https://www.ancestrycdn.com/support/us/2020/08/matchingwhitepaper.pdf
Topor, David, Genealogy testing: Prepare for the emotional reaction, Jun 6 2018, Harvard Health Blog, https://www.health.harvard.edu/blog/genealogy-testing-prepare-for-the-emotional-reaction-2018060613990
Guida-Richards, Melissa, My Half Siblings Found Me On 23andMe. I Wasn’t Prepared For What Happened Next, May 28, 2020, HuffPost, https://www.huffpost.com/entry/discovered-siblings-reunited-23andme-dna-test_n_5e690e55c5b60557280f743e
Kaiser, Molly, I’m 22 and I just met my half sister for the very first time. Here’s how it went, Sep 30, 2022, Today, https://www.today.com/health/essay/dna-test-met-half-sister-rcna49840
Williams, Brianne Kirkpatrick, Watershed DNA, https://www.watersheddna.com/blog
Daniella, I Found My Birth Parents and 7 Half-Siblings Thanks to a MyHeritage DNA Test, Apr 6 2023, MyHeritageBlog, https://blog.myheritage.com/2023/04/i-found-my-birth-parents-and-7-half-siblings-thanks-to-a-myheritage-dna-test/
Imbeault, A DNA test revealed a sister I never knew existed. Now what?, Sep 17 2019, The Globe and the Mail, https://www.theglobeandmail.com/life/first-person/article-a-dna-test-revealed-a-sister-i-never-knew-existed-now-what/
Milligan, Kate, An Only Child’s DNA Surprise, 23andMe Blog, https://blog.23andme.com/articles/an-only-childs-dna-surprise
Molina, ‘Kimberly, My stomach dropped’: Half-sisters find each other through ancestry search, Oct 09, 2018, CBC, https://www.cbc.ca/news/canada/ottawa/half-sisters-discovery-ancestry-dna-1.4849559
Ventura, Risell, Man discovers 18 half-siblings after 23andMe DNA test, Jan 6 2022, 2KUTV, https://kutv.com/news/offbeat/man-discovers-18-half-siblings-after-23andme-dna-test
Hauswirth, Heather, How a DNA test led me to the brother I never knew existed, Nov 14 2018, New York Post, https://nypost.com/2018/11/14/how-a-dna-test-led-me-to-the-brother-i-never-knew-existed/
Segalov, Michael, I took a DNA test and found a new family’: the drama and joy of meeting long-lost relatives, 21 ov 2021, The Guardian, https://www.theguardian.com/global/2021/nov/21/i-took-a-dna-test-and-found-a-whole-new-family
[3] Topor, David, Genealogy testing: Prepare for the emotional reaction, Jun 6 2018, Harvard Health Blog, https://www.health.harvard.edu/blog/genealogy-testing-prepare-for-the-emotional-reaction-2018060613990
[4] Guerrini CJ, Robinson JO, Bloss CC, Bash Brooks W, Fullerton SM, Kirkpatrick B, Lee SS, Majumder M, Pereira S, Schuman O, McGuire AL. Family secrets: Experiences and outcomes of participating in direct-to-consumer genetic relative-finder services. Am J Hum Genet. 2022 Mar 3;109(3):486-497. doi: 10.1016/j.ajhg.2022.01.013. Epub 2022 Feb 24. PMID: 35216680; PMCID: PMC8948156, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8948156/
Lee H, Vogel RI, LeRoy B, Zierhut HA. Adult adoptees and their use of direct-to-consumer genetic testing: Searching for family, searching for health. J Genet Couns. 2021 Feb;30(1):144-157. doi: 10.1002/jgc4.1304. Epub 2020 Jun 29. PMID: 32602181, https://pubmed.ncbi.nlm.nih.gov/32602181/
Roberts JS, Gornick MC, Carere DA, Uhlmann WR, Ruffin MT, Green RC. Direct-to-Consumer Genetic Testing: User Motivations, Decision Making, and Perceived Utility of Results. Public Health Genomics. 2017;20(1):36-45. doi: 10.1159/000455006. Epub 2017 Jan 10. PMID: 28068660, https://pubmed.ncbi.nlm.nih.gov/28068660/
[5] Guerrini CJ, Robinson JO, Bloss CC, Bash Brooks W, Fullerton SM, Kirkpatrick B, Lee SS, Majumder M, Pereira S, Schuman O, McGuire AL. Family secrets: Experiences and outcomes of participating in direct-to-consumer genetic relative-finder services. Am J Hum Genet. 2022 Mar 3;109(3):486-497. doi: 10.1016/j.ajhg.2022.01.013. Epub 2022 Feb 24. PMID: 35216680; PMCID: PMC8948156, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8948156/
[6] Casas KA. Adoptees’ Pursuit of Genomic Testing to Fill Gaps in Family Health History and Reduce Healthcare Disparity. Narrat Inq Bioeth. 2018;8(2):131-135. doi: 10.1353/nib.2018.0050. PMID: 30220696, https://pubmed.ncbi.nlm.nih.gov/30220696/
[7] several DNA testing companies offer communication platforms to connect with genetic matches.
- 23andMe offers a “DNA Relatives” feature where users can contact matches after they agree to share genome.
- Family Tree DNA allows direct email communication with matches6.AncestryDNA provides an internal messaging system for contacting matches.
- MyHeritage uses its own messaging system for match communication.
- Living DNA includes a messaging system to reach out to genetic matches.
Autosomal DNA testing comparison chart, International Society of Genetic Genealogy Wiki, This page was last edited on 8 October 2024, https://isogg.org/wiki/Autosomal_DNA_testing_comparison_chart
[8] McDermott, Marc, How Do Half-Siblings Show Up on Ancestry DNA?, GenealogyExplained, 23 Dec 2022, https://www.genealogyexplained.com/how-do-half-siblings-show-up-on-ancestry-dna/
[9] FIRs (Fully Identical Regions) are genetic segments that are shared between individuals. These regions represent areas of DNA where both chromosomal copies are identical between the compared individuals.
[10] Autosomal DNA Statistics, This page was last edited on 17 October 2022, International Society of Genetic Genealogy Wiki, https://isogg.org/wiki/Autosomal_DNA_statistics
[11] The Shared cM Project (ScP) is a collaborative data collection and analysis project that helps genealogists understand DNA relationships by documenting the ranges of shared centimorgans (cM) associated with various known family relationships. The project contains over 60,000 submissions from genealogists and provides probability estimates for different relationship types based on shared DNA amounts.
Bettinger, Blaine, Version 4.0! March 2020 Update to the Shared cM Project!, 27 Mar 2020, The Genetic Genealogist, https://thegeneticgenealogist.com/2020/03/27/version-4-0-march-2020-update-to-the-shared-cm-project/
Bettinger, Blaine & Jonny Perl, The Shared cM Project 4.0 tool v4, 26 Mar 2020, DNA Painter, https://dnapainter.com/tools/sharedcmv4
Perl, Jonny, Shared cM histograms: did you know? #3, 12 Apr 2023, DNA Painter Blog, https://blog.dnapainter.com/blog/shared-cm-histograms-did-you-know-3/
Shared cM | How Am I Related to My DNA Matches?, Your DNA Guide, https://www.yourdnaguide.com/shared-cm-project
[12] The cM test results for matches can differ between DNA companies. For example the table reflects the estimates cM values for matches between me and my half brothers based on AncestryDNA and 23andMe test results.
These cM values are based on converting the percentage of shared cM values obtained in the 23andMe atDNA test results. Since 23andMe only provides percent of shared cMs between me and Dave or greg, you need to use a conversion procedure:
There are two ways to convert 23andMe matches to centimorgans (cM), you can use the Shared cM Project tool at DNA Painter:
- Go to the Shared cM Project tool at DNA Painter
- Enter the percentage of shared DNA in the percentage box
- The tool will show you the cMs
Bettinger, Blaine,, The Shared cM Project 4.0 Tool v4, Mar 2020, DNA Painter, https://dnapainter.com/tools/sharedcmv4
You can also use a ‘quick and dirty’ approach to convert the percentage into centimorgans by just multiplying your percentage by 68.
Cooke, Lisa, What’s a CentiMorgan, Anyway? How DNA Tests for Family History Measure Genetic Relationships, 23 Oct 2017, Genealogy Gems, https://lisalouisecooke.com/2017/10/23/genetic-relationships-centimorgans/
Fully identical region, This page was last edited on 1 April 2022, International Society of Genetic Genealogy Wiki, https://isogg.org/wiki/Fully_identical_region
Estes, Roberta, Pedigree Collapse and DNA – Plus an Easy-Peasy Shortcut, 31 Jan 2024, DNAeXplained – Genetic Genealogy, https://dna-explained.com/category/fully-identical-regions/
Hill-Burns, Erin, How much DNA in FIRS(Fully Identical Regions) do relatives share?, Genes & History, https://genesandhistory.wordpress.com/2019/12/04/how-much-dna-in-firs-fully-identical-regions-do-relatives-share/
DNA Geek, AncestryDNA Is Using FIRs to Distinguish Full and Half Siblings, 7 Feb 2019, TheDNAGeek, https://thednageek.com/ancestrydna-is-using-firs-to-distinguish-full-and-half-siblings/
SegcM | DNA Science, Relationship predictions that use both the # of segments and total cMs https://dna-sci.com/tools/segcm/
DNA-Sci, Segments Matter! , 3 Feb 2023, DNA Science Blog, https://dna-sci.com/2023/02/03/segments-matter/
[13] McDermott, Marc, How Do Half-Siblings Show Up on Ancestry DNA?, GenealogyExplained, 23 Dec 2022, https://www.genealogyexplained.com/how-do-half-siblings-show-up-on-ancestry-dna/
What is the best test for showing that two people are half siblings? 7 Jan 2016, The Tech Interactive, https://www.thetech.org/ask-a-geneticist/articles/2016/best-half-sibling-dna-test/
Estes, Roberta, Full or Half Siblings?, 3 Apr 2019, DNAeXplained – Genetic Genealogy, https://dna-explained.com/2019/04/03/full-or-half-siblings/
[14] McDermott, Marc, How Do Half-Siblings Show Up on Ancestry DNA?, GenealogyExplained, 23 Dec 2022, https://www.genealogyexplained.com/how-do-half-siblings-show-up-on-ancestry-dna/
Stocker CM, Gilligan M, Klopack ET, Conger KJ, Lanthier RP, Neppl TK, O’Neal CW, Wickrama KAS. Sibling relationships in older adulthood: Links with loneliness and well-being. J Fam Psychol. 2020 Mar;34(2):175-185. doi: 10.1037/fam0000586. Epub 2019 Aug 15. PMID: 31414866; PMCID: PMC7012710. https://pmc.ncbi.nlm.nih.gov/articles/PMC7012710/
Segments Matter!
[15] Baby Scoop Era, Wikipedia, This page was last edited on 22 October 2024,, https://en.wikipedia.org/wiki/Baby_Scoop_Era
[16] Solinger, Rickie, Wake Up Susie: Single Pregnancy and Race Before Roe v. Wade, New York: Routledge, 2000, Page 102
[17] The Comstock Act of 1873 severely restricted access to birth control in the United States through several key measures. It criminalized mailing or distributing any contraceptive devices or information about contraception. Imposed harsh penalties including fines of $100-$5,000 and imprisonment of 1-10 years for violations. Led to thousands of arrests and the destruction of hundreds of tons of books and educational materials about contraception.
The Comstock Act prevented women from accessing information about their reproductive health and pregnancy prevention options. It banned doctors and social reformers from providing contraceptive information to patients. State-level “Comstock laws” further expanded restrictions on contraception, with some states like Connecticut completely banning birth control use. The Comstock Act’s restrictions on contraception remained technically in effect until 1971, when Congress finally removed the language related to contraceptives from the law.
Wexler, Ellen, The 150-Year-Old Comstock Act Could Transform the Abortion Debate, 15 Jun 2023, Smithsonian Magazine, https://www.smithsonianmag.com/history/comstock-act-transform-abortion-debate-180982363/
Comstock act, Women & the American Story, The New York Historical, https://wams.nyhistory.org/industry-and-empire/fighting-for-equality/comstock-act/
Birth control in the United States, Wikipedia, This page was last edited on 12 November 2024, https://en.wikipedia.org/wiki/Birth_control_in_the_United_States
Comstock Act of 1873 Wikipedia, This page was last edited on 15 November 2024, https://en.wikipedia.org/wiki/Comstock_Act_of_1873
[18] A Timeline of Contraception, American Experience, PBS, https://www.pbs.org/wgbh/americanexperience/features/pill-timeline/
Birth control in the United States, Wikipedia, This page was last edited on 12 November 2024, https://en.wikipedia.org/wiki/Birth_control_in_the_United_States
[19] Gibson, Megan, One Factor That Kept the Women of 1960 Away From Birth Control Pills: Cost, 23 Jun 2015, Time, https://time.com/3929971/enovid-the-pill/
See also:
A Timeline of Contraception, American Experience, PBS, https://www.pbs.org/wgbh/americanexperience/features/pill-timeline/
From Acacia to IUDs: The History of Birth Control in the United States, HealthLine, https://www.healthline.com/health/birth-control/history-of-birth-control
[20] Gold, Rachel Benson, Lessons from Before Roe: Will Past be Prologue?, Volume 6, Issue 1, Guttmacher Policy Review, 1 Mar 2003, https://www.guttmacher.org/gpr/2003/03/lessons-roe-will-past-be-prologue
Paintin, D. (1998). A Medical View of Abortion in the 1960s. In: Lee, E. (eds) Abortion Law and Politics Today. Palgrave Macmillan, London. https://doi.org/10.1007/978-1-349-26876-4_2
[21] Herman, Ellen. “The Paradoxical Rationalization of Modern Adoption.” Journal of Social History, 36, no. 1 (Winter 2002): 339-385.
Herman, Ellen. Kinship by Design: A History of Adoption in the Modern United States of America. Chicago: University of Chicago Press, 2008.
[22] Owens, Rudy, Number of Adoptees Relinquished: 1944-1975, , You Don’t Know How Lucky You are, https://www.howluckyuare.com/numbers-adoptees-relinquished-1944-1975/
[23] National Florence Crittenton Mission, Wikipedia, This page was last edited on 17 February 2024, https://en.wikipedia.org/wiki/National_Florence_Crittenton_Mission
Florence Crittenton Mission, VCU Libraries, Social Welfare History Project, Virginia Commonwealth University, https://socialwelfare.library.vcu.edu/programs/child-welfarechild-labor/florence-crittenton-mission/
[24] Illegitimate Births in Vital Statistics of the United States,1960, Volume I – Natality, Pages l-12 and l-13. https://www.cdc.gov/nchs/data/vsus/nat60_1.pdf
Penelope L. Maza, “Adoption Trends: 1944-1975”, Child Welfare Research Notes No. 9, U.S. Children’s Bureau, August 1984
Franks, Julia, The American History Behind the Novel ‘The Say So’, Illegitimate Pregnancies, http://www.juliafranks.com/the-say-so-the-history
See also:
Bernstein, Rose. “Unmarried Parents,” Encyclopedia of Social Work. Issue 5. New York National Association of Social Workers, 1965, p. 797
Shlakman, Vera. “Unmarried Parenthood: An Approach to Social Policy.” Social Casework, vol. 42, October 1966, p. 494
Solinger, Rickie. Wake Up Little Susie: Single Pregnancy and Race Before Roe V. Wade, 2nd edition, Routledge, 2000
Moriguchi, Chiaki. (2012). The Evolution of Child Adoption in the United States, 1950-2010: An Economic Analysis of Historical Trends, Discussion Paper Series A No.572, June 2012, https://www.researchgate.net/publication/254420379_The_Evolution_of_Child_Adoption_in_the_United_States_1950-2010_An_Economic_Analysis_of_Historical_Trends
[25] Franks, Julia, The American History Behind the Novel ‘The Say So’, Illegitimate Pregnancies, http://www.juliafranks.com/the-say-so-the-history
[26] Owens, Rudy, Number of Adoptees Relinquished: 1944-1975, , You Don’t Know How Lucky You are, https://www.howluckyuare.com/numbers-adoptees-relinquished-1944-1975/



{kind=link}